

 下载:
下载:
-
RRx-001是一种源于航空工业的肿瘤免疫治疗药物[1],其化学结构中含有溴代乙酰和双硝基氮杂环丁基团,目前处于III期临床试验[2]。RRx-001作为单一药物、放疗增敏剂或免疫增敏剂[3],用于细胞肺癌[4]、转移性结直肠癌[5]、卵巢癌[6]和胶质母细胞瘤[7]等肿瘤的治疗,显示出良好的疗效和安全性。研究发现,RRx-001具有多种作用机制,如靶向CD47-SIRPα信号通路,使肿瘤相关巨噬细胞复极化,由抗炎症M2表型转为促炎症M1表型[8];使肿瘤血管正常化,增加化疗药物渗透,产生代谢产物RONS,导致肿瘤细胞坏死[9];通过表观遗传抑制活性激活抑癌基因[10]等。作为靶向CD47的小分子药物,RRx-001在临床试验中未发现CD47抗体药物常见的嗜血综合征,表现出优于抗体大分子药物的安全性[11]。但RRx-001也存在明显的输注部位反应,因而临床需要采用特殊的注射器材,通过血液共同输注方式给药。
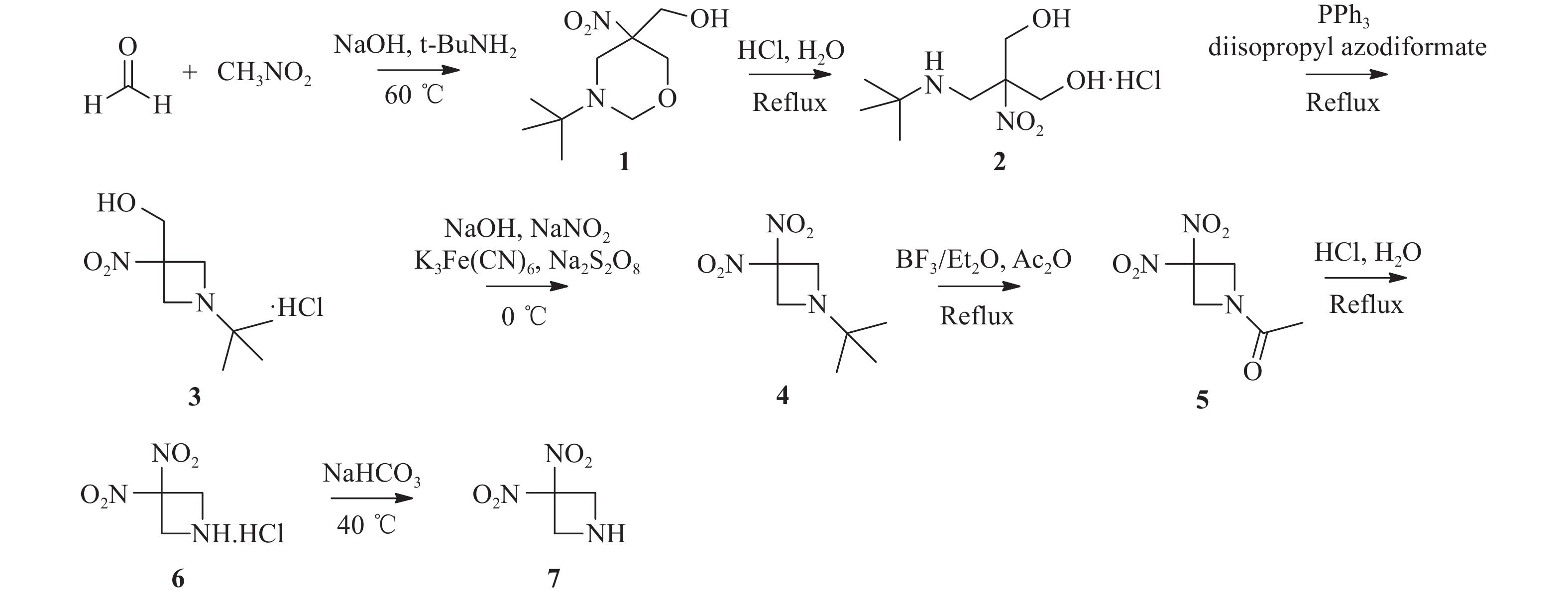
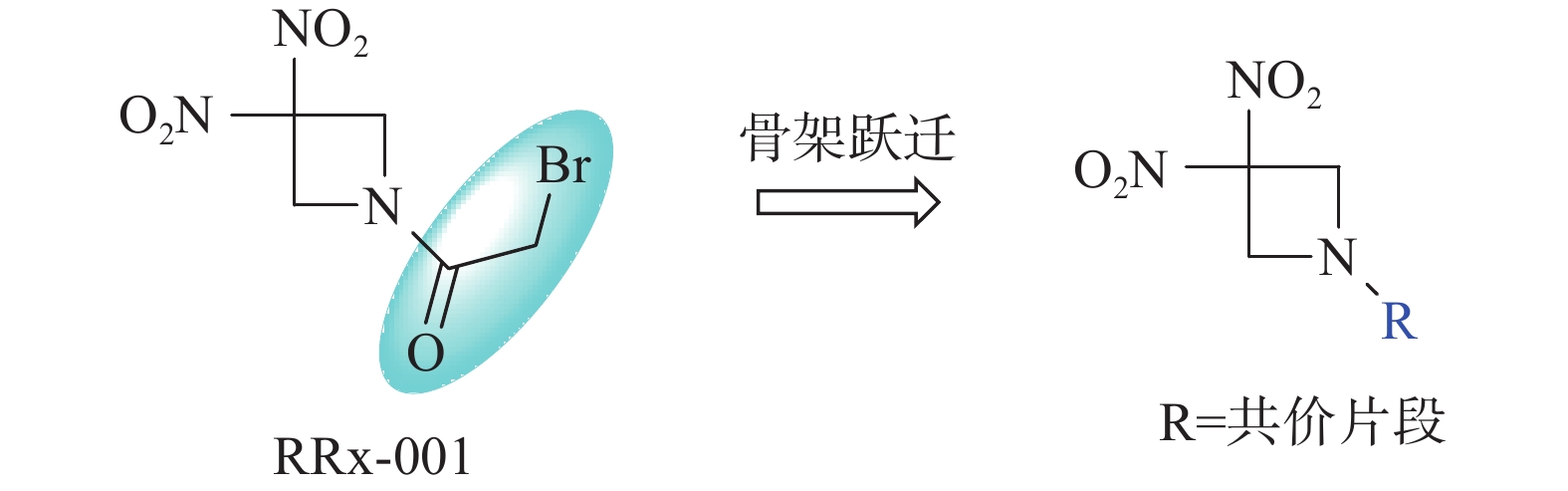
RRx-001结构中的溴代乙酰基为共价结合片段,也是其产生活性的药效基团[1]。为进一步探讨RRx-001的构效关系,本文拟采用骨架跃迁的药物设计策略,以不同的共价结合片段代替溴代乙酰基,分析其对抗肿瘤活性的影响,从而为后续基于RRx-001骨架的药物设计研究提供借鉴(图1)。
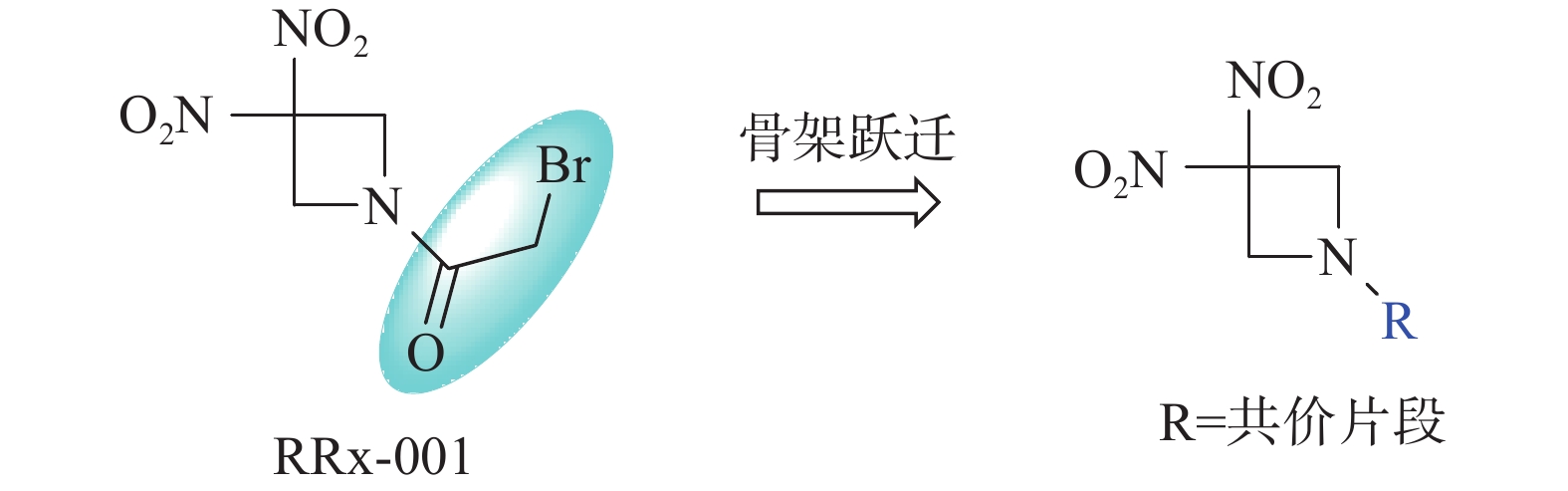
图 1 RRx-001化学结构及其衍生物设计策略
-
实验所用试剂分别采购自泰坦、毕得、乐研等公司,各类试剂均为市售分析纯或化学纯。核磁共振仪采用的是德国Bruke的600、400和300 MHz型,采用TMS作为内标,DMSO-d6、CDCl3或D2O等为溶剂。耦合常数(J)和化学位移(δ)单位分别用Hz和ppm来表示。高分辨质谱为德国Bruke micrOTOF
10257 ,抗肿瘤活性测试所用仪器为Biotek Synergy H2多功能酶标仪。薄层色谱(TLC)使用的硅胶板为GF254(中国青岛海洋化学),柱层析使用的是200~300目硅胶(中国青岛海洋化学)。 -
将多聚甲醛(24.1 g,0.8 mol)加入250 ml三颈烧瓶中,然后加入0.16 %氢氧化钠水溶液(40 ml),升温至40 ℃。缓慢滴加硝基甲烷(10.6 ml,0.2 mol),滴加结束后升温至60 ℃,滴加叔丁胺(21.2 ml,0.2 mol),滴加结束后继续反应10 min。反应液冷却至室温后过滤,水洗,干燥得38.8 g白色固体1,收率89.4 %,mp:136.1~138.2 ℃。1H NMR(400 MHz, DMSO-d6)δ:5.35(t, J=5.8 Hz, 1 H),4.42(t, J=10.0 Hz, 2 H),3.81(d, J=7.9 Hz, 1 H),3.63–3.51(m, 4 H),2.59(d, J=12.3 Hz, 1 H),0.95(s, 9 H)。
-
两颈烧瓶中依次加入500 ml无水乙醇、12 ml浓盐酸和化合物1(20.0 g,0.09 mol),加热回流反应6 h。反应结束后,减压蒸去溶剂,加入异丙醇搅拌0.5 h,过滤,洗涤,干燥得17.0 g白色晶体2,收率78.2 %,mp:175.5~178.3 ℃。1H NMR(400 MHz, D2O)δ:4.12(d, J=12.5 Hz, 2 H), 3.88(d, J=12.5 Hz, 2 H), 3.76(s, 2H), 1.36(s, 9 H)。
-
在氮气保护下,将化合物2(5.0 g,0.02 mol)加入到250 ml三颈烧瓶中,分别加入50 ml无水四氢呋喃和偶氮二甲酸二异丙酯(5.6 g,0.03 mol),升温至60 ℃。然后,将溶有三苯基磷(7.4 g,0.03 mol)的四氢呋喃溶液(15 ml)滴加到反应液中,滴加结束后继续反应5 h。反应液冷却至室温后过滤,洗涤,干燥得3.9 g白色固体3,收率86.6 %,mp:165.3~174.0 ℃。1H NMR(400 MHz, D2O)δ:4.74(s, 2 H), 4.43(s, 2 H), 4.16(s, 2 H), 1.26(s, 9 H)。
-
将化合物3(7.0 g,0.03 mol)和氢氧化钠水溶液(4.3 g,90 ml H2O)加入到250 ml两颈烧瓶中,室温搅拌2 h,冰浴冷却到10 ℃以下后,滴加亚硝酸钠(8.6 g,0.12 mol)、K3Fe(CN)6(1.0 g,0.003 mol)和水(5 ml)配成的溶液。控制反应温度为10~15 ℃下,分批次加入过硫酸钠(10.3 g,0.04 mol),然后升温至室温,反应过夜。反应液用20 ml二氯甲烷萃取3次,有机相用无水硫酸钠干燥,过滤,滤液减压蒸去溶剂得5.5 g黄色液体4,收率88.1 %。1H NMR(400 MHz, CDCl3)δ:4.12(s, 4 H), 1.05(s, 9 H)。
-
25 ml两颈烧瓶中依次加入化合物4(5.0 g,0.03 mol)、醋酸酐(9.8 ml,0.11 mol)和三氟硼酸乙醚溶液(0.5 ml,3.88 mmol),加热至120 ℃,回流反应12 h。减压蒸去溶剂,柱层析纯化(二氯甲烷 : 甲醇=100 : 1)得2.8 g淡黄色固体5,收率60.0 %,mp:110.1~113.9 ℃。1H NMR(600 MHz, CDCl3)δ:4.98(s, 2 H), 4.82(s, 2 H), 2.02(s, 3 H)。
-
将化合物5(1.0 g,5.29 mmol)加入到50 ml单颈烧瓶,然后加入6.3 ml 的5 %盐酸溶液,回流反应4 h。反应液冷却至室温,过滤,滤液减压蒸去溶剂得0.6 g黄色固体6,收率58.2 %,mp:150.3~155. 2 ℃。1H NMR(600 MHz, DMSO-d6)δ:4.98(s, 4 H), 3.42(s, 2 H)。
-
将化合物6(0.6 g,3.16 mmol)和15 ml水加入到50 ml两颈烧瓶中,升温至40 ℃,再滴加5 %碳酸氢钠溶液至pH为8。反应液用10 ml二氯甲烷萃取3次,有机相用无水硫酸钠干燥,过滤,减压蒸去溶剂得0.4 g黄色油状液体7,收率84.7 %。1H NMR(600 MHz, CDCl3)δ:4.51(s, 4 H), 2.20(s, 1 H)。
-
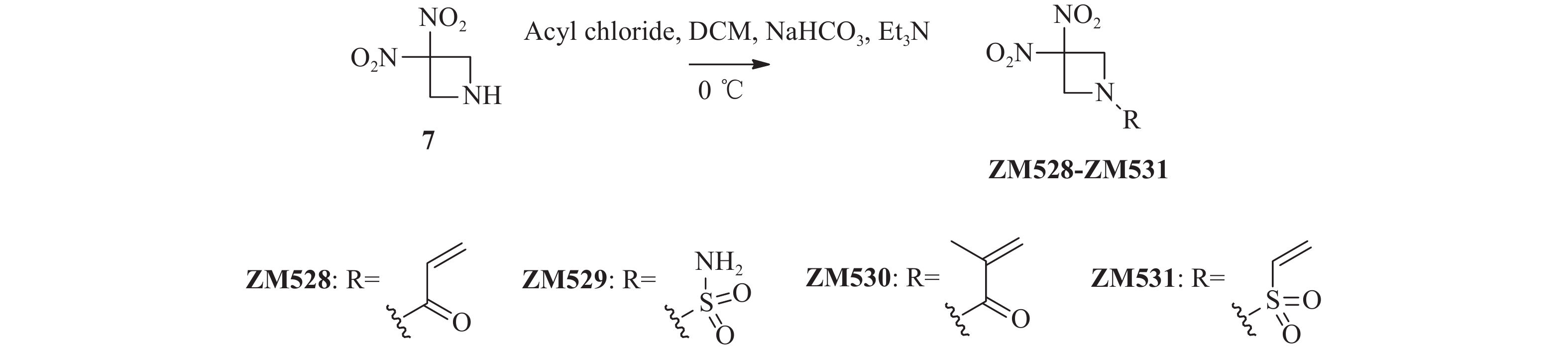
将关键中间体3, 3-二硝基氮杂环丁烷7(74.0 mg,0.50 mmol)加入到25 ml单颈烧瓶后,分别加入干燥的二氯甲烷(4 ml)和碳酸氢钠(42.0 mg,0.50 mmol),冰浴冷却至0 ℃后,滴加各种酰氯或磺酰氯(0.55 mmol),完全反应后减压蒸去溶剂,柱层析纯化(二氯甲烷 : 甲醇=100 : 1)得化合物ZM528~ZM531。
ZM528,白色固体,83 mg,收率61.3%。1H NMR(300 MHz, CDCl3) δ: 6.46(dd, J=16.9, 1.2 Hz, 1 H), 6.16(dd, J=16.9, 10.4 Hz, 1 H), 5.88(dd, J=10.4, 1.2 Hz, 1 H), 4.96(s, 4 H). HRMS(ESI, positive)m/z calcd for C5H7N3O5 [M + H]+:
202.0464 ; found 202.0458。ZM529,白色固体,77 mg,收率60.2%。1H NMR(300 MHz, DMSO-d6)δ: 7.47(s, 2 H), 4.77(s, 4 H). HRMS(ESI, positive)m/z calcd for C3H6N4O6S [M + H]+:
226.0008 ; found 226.0013。ZM530,白色固体,54 mg,收率50.1%。1H NMR(600 MHz, CDCl3)δ: 5.63–5.61(m, 1 H), 5.45(s, 1 H), 4.93(s, 4 H), 1.97(dd, J=1.6, 1.1 Hz, 3 H)。HRMS(ESI, positive)m/z calcd for C7H9N3O5 [M + H]+:
216.0620 ; found 216.0615。ZM531,淡黄色固体,81 mg,收率69.6%。1H NMR(600 MHz, CDCl3)δ: 6.58(dd, J=16.5, 9.8 Hz, 1 H), 6.46(d, J=16.6 Hz, 1 H), 6.27(d, J=9.8 Hz, 1 H), 4.75(s, 4 H)。
-
选用人结肠癌细胞HCT-116和人非小细胞肺癌细胞A549,于海军军医大学药物化学教研室冻存和传代。96孔板边缘每孔加入100 μl的PBS溶液防止边缘效应,内部每孔加入浓度为7×104个/ml的细胞悬液100 μl,置于37 ℃、5%二氧化碳培养箱内。24 h后,弃去96孔板内培养液,每孔分别加入100 μl受试化合物样品液和对照品液,设三复孔。将96孔板置于37 ℃、5%二氧化碳培养箱中培养72 h。实验采用CCK-8法[14-16]。在基础培养基中加入10 % CCK-8试剂制成混合液,弃去96孔板内旧培养基,加入混合液,100 μl/孔,将96孔板置于37 ℃、5 %二氧化碳培养箱中孵育2~4 h。使用酶标仪于450 nm波长处测定荧光OD值。细胞生长抑制率IC%=(空白对照孔OD值-给药孔OD值)/空白对照孔OD值×100%。根据各个浓度的IC%值,用GraphPad软件进行线性回归,算出各受试化合物抑制细胞生长50%的药物浓度,即IC50。
-
参考文献[12-13]设计了关键中间体3, 3-二硝基氮杂环丁烷的合成路线,合成路线见图2。以多聚甲醛和硝基甲烷为起始原料,通过环合、开环、Mistunobu、反向Henrry、脱叔丁基/乙酰化、脱乙酰基、碱化等7步反应合成出关键中间体7,总收率为14.9 %。其中,反向Henrry反应对催化剂的用量和反应温度较为敏感,经过反应条件优化,反应温度为10~15 ℃时,亚硝酸钠和K3Fe(CN)6的用量分别为化合物3的4倍和10%时,该步反应收率达88.1%,相比原有路线提升了6.3%。
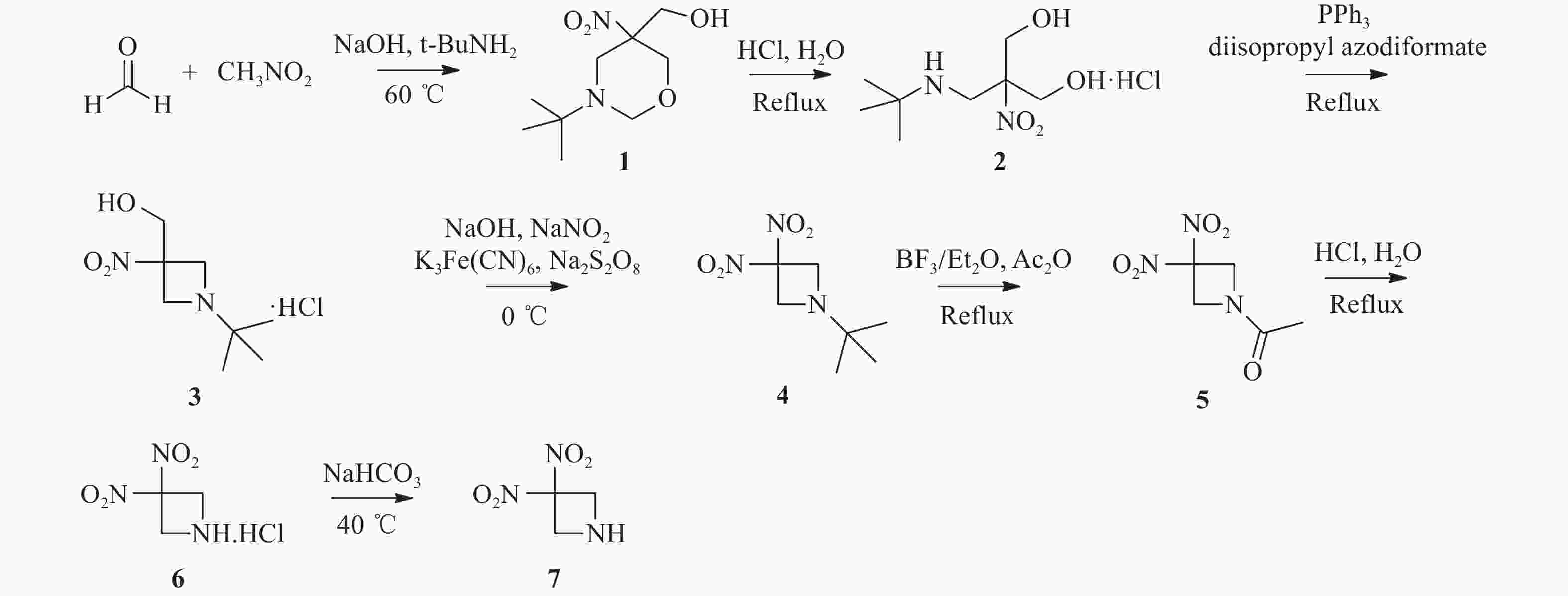
图 2 关键中间体3, 3-二硝基氮杂环丁烷的合成路线
将关键中间体7与各种酰氯或磺酰氯在三乙胺和碳酸氢钠催化下发生酰胺化反应,以较高收率合成得到目标化合物ZM528~ZM531(图3)。
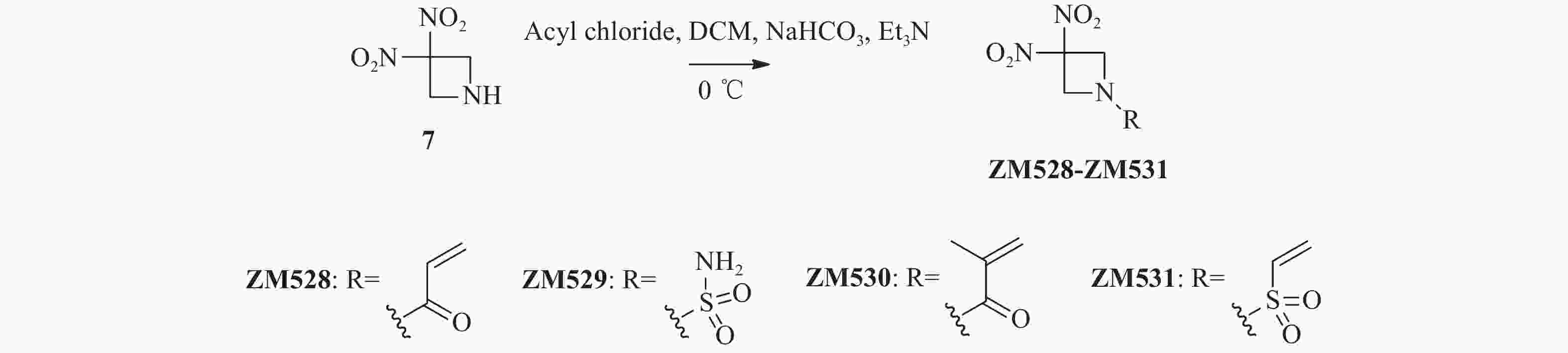
图 3 RRx-001类似物的合成路线
-
采用CCK-8法测定目标化合物对肿瘤细胞株HCT-116和A549的体外抗肿瘤活性,以多柔比星(DOX)和RRx-001为阳性对照药,结果见表1。从表中可以看出,4个化合物均能保持一定的抗肿瘤活性,但相比RRx-001,均出现明显下降,其中丙烯酰基类化合物ZM528活性最高,对HCT-116和A549的IC50分别为(6.0±2.7)和(5.1±4.8)μmol/L。当丙烯酰基的2位引入甲基后活性明显下降,以磺酰基代替酰基后,活性也呈现下降趋势。以磺酰胺基代替丙烯酰基后,对HCT-116和A549肿瘤细胞株的活性分别下降了6.4和5.4倍,但当以磺酰基代替磺酰胺基后活性基本保持。以上研究结果初步表明,RRx-001的溴代乙酰基对抗肿瘤活性影响较大,以其他共价结合片段代替后活性出现明显下降。
表 1 RRx-001衍生物体外抗肿瘤活性
化合物IC50(μmol/L) HCT-116细胞株 A549细胞株 RRx-001 0.94±0.7 <0.2 ZM528 6.0±2.7 5.1±4.8 ZM529 38.1±19.7 27.3±10.4 ZM530 >50 14.4±3.0 ZM531 34.5±13.5 36.3±5.3 DOX <0.2 <0.2 -
采用骨架跃迁药物设计策略,设计合成出4个RRx-001衍生物。体外抗肿瘤活性研究发现,所有新化合物对HCT-116和A549细胞株的抑制活性均显示出明显的下降,但能保持一定的抗肿瘤活性,其中丙烯酰基类化合物ZM528活性最高,对两种肿瘤细胞株的IC50分别为(6.0±2.7)和(5.1±4.8) μmol/L,其原因可能是RRx-001的共价结合片段为溴乙酰基,结合活性高于其他共价结合片段。初步构效关系显示,氮杂环丁烷的氨基上溴代乙酰基取代后活性最高,丙烯酰基取代活性次之。研究结果和初步构效关系为以RRx-001为骨架的新型靶向CD47药物的进一步优化设计研究提供了理论指导。
Synthesis and Antitumor Activity of Novel RRx-001 Derivatives
-
摘要:
目的 研究引入共价结合片段后的RRx-001衍生物的抗肿瘤活性。 方法 设计合成了4个目标化合物,其结构通过1H NMR和HRMS等确证;选择人肺癌细胞株A549和人结肠癌细胞株HCT116进行体外抗肿瘤活性测试。 结果 所有化合物均显示出一定的抗肿瘤活性,其中化合物 ZM528 活性最高,对两种肿瘤细胞株的IC50值分别为(5.1±4.8)和(6.0±2.7)μmol/L。 结论 以新的共价结合片段代替RRx-001的溴代乙酰基后可保持抗肿瘤活性。 Abstract:Objectives To study the antitumor activities of RRx-001 derivatives with novel covalent fragments Methods Four targeted compounds were designed and synthesized. The structures were confirmed by 1H NMR and HRMS. A549 and HCT116 cancer cell lines were selected for antiproliferative activity assays. Results All the compounds revealed antitumor activities and compound ZM528 showed the best antitumor activity against these two cell lines with IC50 values of (5.1±4.8) and (6.0±2.7) μmol/L, respectively. Conclusions The result indicated that bromoacetyl group of RRx-001 could be substituted with other covalent fragments. -
Key words:
- RRx-001 /
- immuno-oncology /
- covalent inhibitor /
- synthesis /
- antitumor
-
表 1 RRx-001衍生物体外抗肿瘤活性
化合物IC50(μmol/L) HCT-116细胞株 A549细胞株 RRx-001 0.94±0.7 <0.2 ZM528 6.0±2.7 5.1±4.8 ZM529 38.1±19.7 27.3±10.4 ZM530 >50 14.4±3.0 ZM531 34.5±13.5 36.3±5.3 DOX <0.2 <0.2  下载: 导出CSV
下载: 导出CSV
-
[1] ORONSKY B, GUO X N, WANG X H, et al. Discovery of RRx-001, a myc and CD47 downregulating small molecule with tumor targeted cytotoxicity and healthy tissue cytoprotective properties in clinical development[J]. J Med Chem, 2021, 64(11):7261-7271. doi: 10.1021/acs.jmedchem.1c00599 [2] ORONSKY B, REID T R, LARSON C, et al. REPLATINUM Phase III randomized study: RRx-001 + platinum doublet versus platinum doublet in third-line small cell lung cancer[J]. Future Oncol, 2019, 15(30):3427-3433. doi: 10.2217/fon-2019-0317 [3] JURGENSEN K J, SKINNER W K J, ORONSKY B, et al. RRx-001 radioprotection: enhancement of survival and hematopoietic recovery in gamma-irradiated mice[J]. Front Pharmacol, 2021, 12:676396. doi: 10.3389/fphar.2021.676396 [4] MORGENSZTERN D, ROSE M, WAQAR S N, et al. RRx-001 followed by platinum plus etoposide in patients with previously treated small-cell lung cancer[J]. Br J Cancer, 2019, 121(3):211-217. doi: 10.1038/s41416-019-0504-8 [5] REID T R, ABROUK N, CAROEN S, et al. ROCKET: phase II randomized, active-controlled, multicenter trial to assess the safety and efficacy of RRx-001 + irinotecan vs. single-agent regorafenib in third/fourth line colorectal cancer[J]. Clin Colorectal Cancer, 2023, 22(1):92-99. doi: 10.1016/j.clcc.2022.11.003 [6] REID T, ORONSKY B, CAROEN S, et al. Phase 1 pilot study of RRx-001 + nivolumab in patients with advanced metastatic cancer(PRIMETIME)[J]. Front Immunol, 2023, 14:1104753. doi: 10.3389/fimmu.2023.1104753 [7] FINE H, REID T, CAROEN S, et al. A multicenter, phase 1, dose escalation clinical trial(G-FORCE-1)of XRT, RRx-001 and temozolomide followed by temozolomide +/- RRx-001 in newly diagnosed glioblastoma[J]. Front Oncol, 2023, 13:1176448. doi: 10.3389/fonc.2023.1176448 [8] ORONSKY B, PAULMURUGAN R, FOYGEL K, et al. RRx-001: a systemically non-toxic M2-to-M1 macrophage stimulating and prosensitizing agent in Phase II clinical trials[J]. Expert Opin Investig Drugs, 2017, 26(1):109-119. doi: 10.1080/13543784.2017.1268600 [9] JANI V P, ASARO R, ORONSKY B, et al. RRx-001 increases erythrocyte preferential adhesion to the tumor vasculature[J]. Int J Mol Sci, 2021, 22(9):4713. doi: 10.3390/ijms22094713 [10] ZHAO H J, NING S C, SCICINSKI J, et al. Epigenetic effects of RRx-001: a possible unifying mechanism of anticancer activity[J]. Oncotarget, 2015, 6(41):43172-43181. doi: 10.18632/oncotarget.6526 [11] ORONSKY B, CABRALES P, CAROEN S, et al. RRx-001, a downregulator of the CD47- SIRPα checkpoint pathway, does not cause anemia or thrombocytopenia[J]. Expert Opin Drug Metab Toxicol, 2021, 17(4):355-357. doi: 10.1080/17425255.2021.1876025 [12] 李洪珍, 舒远杰, 刘世俊, 等. N-乙酰基-3, 3-二硝基氮杂环丁烷的合成[J]. 化学研究与应用, 2004, 16(3):393-395. [13] 李洪珍, 舒远杰, 黄奕刚, 等. 3, 3-二硝基氮杂环丁烷和1, 1'-亚甲基-双(3, 3-二硝基-1-氮杂环丁烷)的合成研究[J]. 有机化学, 2004, 24(7):775-777. [14] TANG W M, ZHANG Y M, YANG K L, et al. Discovery of novel 3, 11-bispeptide ester arenobufagin derivatives with potential in vivo antitumor activity and reduced cardiotoxicity[J]. Chem Biodivers, 2023, 20(2):e202200911. doi: 10.1002/cbdv.202200911 [15] ZHANG Y M, YANG K L, YE S, et al. Application of a fluorine strategy in the lead optimization of betulinic acid to the discovery of potent CD73 inhibitors[J]. Steroids, 2022, 188:109112. doi: 10.1016/j.steroids.2022.109112 [16] 罗川, 马建江, 缪震元, 等. 沙蟾毒精酯类衍生物的合成和抗肿瘤活性研究[J]. 药学实践杂志, 2021, 39(1):35-37,57. -
 点击查看大图
点击查看大图
图(3) / 表(1)
计量
- 文章访问数: 226
- HTML全文浏览量: 113
- PDF下载量: 1
- 被引次数: 0
