

 下载:
下载:
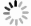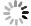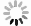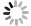
-
NLRP3(NLR family pyrin domain containing 3)炎症小体作为固有免疫的重要组分,在机体免疫反应、炎症性疾病发生和发展过程中发挥着重要作用。NLRP3炎症小体包括传感器(NLRP3受体蛋白)、适配器(含有CARD的凋亡相关斑点样蛋白)和效应器(caspase-1蛋白),其中,NLRP3受体蛋白是炎症反应中的关键调控蛋白[1]。研究发现,NLRP3炎症小体的激活可以分为经典激活途径和非经典激活途径,在NLRP3炎症性小体的经典激活途径中,激活全过程可以分为启动和激活两阶段。NLRP3炎症小体的激活在先天免疫反应调节中起着关键作用,但过度激活会导致自身免疫性疾病、中枢神经系统疾病、代谢性疾病、心血管疾病和癌症等多种疾病的发生和进展[2-4]。因此,以NLRP3炎症小体为靶点的药物研发成为药物设计领域的热点[5-6]。
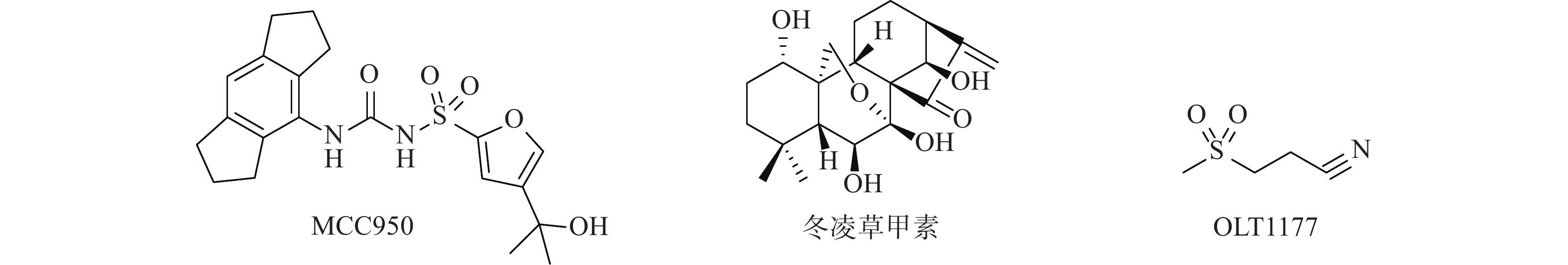
迄今为止报道的NLRP3抑制剂可分为直接抑制NLRP3炎症小体激活的抑制剂和通过抑制炎症小体成分或相关信号通路来间接抑制NLRP3炎症小体激活的抑制剂(图1)[7-8]。Canakinumab是诺华制药公司研发的一种能抑制IL-1β活性的抗IL-1β单克隆抗体,在2009年获得美国食品药物管理局批准用于对冷吡啉相关周期性综合征患者的治疗[9]。MCC950是由辉瑞公司研发的二芳基磺酰脲类NLRP3炎性小体抑制剂,目前已进入临床研究。MCC950能直接与NLRP3受体蛋白NACHT结构域的Walker B基序产生非共价结合,使NLRP3炎症小体处于非活性状态,抑制NLRP3活化和ASC寡聚,阻断NLRP3 炎症小体的经典和非经典激活途径[10-11]。此外,还有多个小分子抑制剂也进入了临床试验阶段[12]。
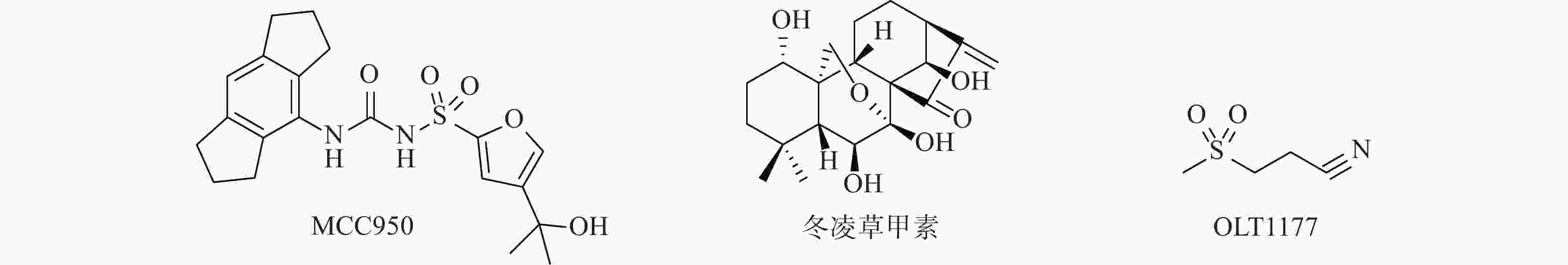
图 1 代表性NLRP3炎症小体小分子抑制剂的化学结构
天然产物是新药研发中先导化合物的重要来源,冬凌草甲素是从中药材冬凌草中提取分离得到,具有抗肿瘤、抗炎、抗菌等多种药理活性的四环二萜类化合物。研究发现,冬凌草甲素通过靶向NLRP3受体蛋白的Cys279来阻断NLRP3和NEK7之间的相互作用,特异性抑制NLRP3炎症小体的激活。其中,冬凌草甲素中的迈克尔受体片段被认为是产生活性的关键药效团[13-15]。为进一步研究冬凌草甲素的抗炎活性构效关系,研发基于天然产物冬凌草甲素的新型NLRP3炎症小体抑制剂,本课题组采用光催化化学反应[16]和骨架跃迁药物设计策略,设计合成出一类冬凌草甲素磺酰脲类衍生物,并考察其抗炎活性。
-
实验所用试剂分别采购自泰坦、毕得、乐研等公司,各类试剂均为市售分析纯或化学纯。600 MHz型核磁共振波谱仪(德国 Bruke 公司),采用 TMS 作为内标,使用 DMSO-d6或D2O 等为溶剂。耦合常数(J)和化学位移(δ)单位分别用 Hz 和 ppm 表示。高分辨质谱采用的是德国 Bruke micrOTOF 10257。薄层色谱(TLC)使用的硅胶板为 GF254(中国青岛海洋化学),柱层析使用的是 200 ~ 300目硅胶(中国青岛海洋化学)。
-
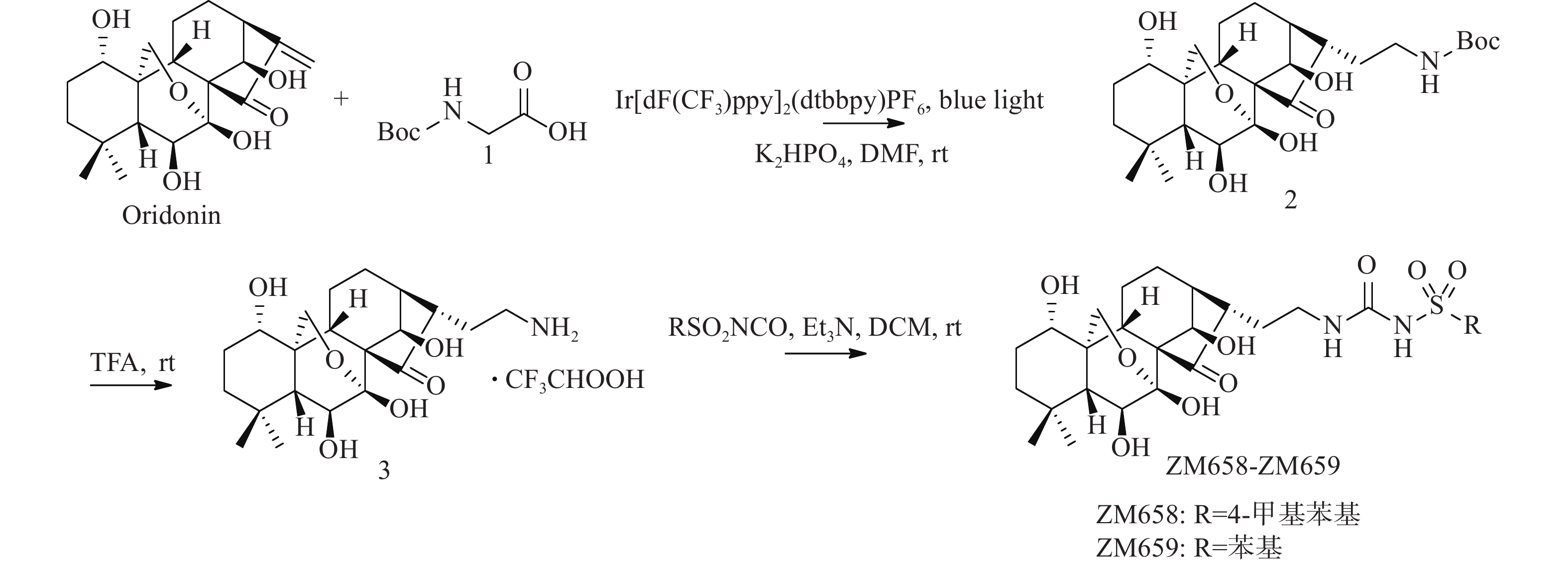
将冬凌草甲素(473 mg,1.20 mmol)、N-Boc-甘氨酸(252 mg,1.44 mmol)光催化剂Ir[dF(CF3)ppy]2(dtbbpy)PF6(14 mg,0.012 mmol)和DMF(6 ml)加入到10 ml玻璃瓶中。然后,加入磷酸氢二钾(251 mg,1.44 mmol),密封玻璃瓶,脱气15 min后充入氮气,在蓝光LED灯带照射下室温反应36 h。反应结束后加入10 ml水,乙酸乙酯萃取3次(10 ml × 3)。合并有机相,水洗(10 ml),无水Na2SO4干燥,减压蒸去溶剂,柱色谱纯化得507 mg黄色固体2(二氯甲烷∶甲醇=100∶3),收率85.0%。1H NMR(600 MHz, DMSO-d6)δ 6.87(t, J = 5.5 Hz, 1 H), 6.78(s, 1 H), 6.08(d, J = 2.1 Hz, 1 H), 5.71(d, J = 10.9 Hz, 1 H), 4.80(s, 1 H), 4.35(d, J = 5.0 Hz, 1 H), 4.10(d, J = 5.0 Hz, 1 H), 4.07(d, J = 10.1 Hz, 1 H), 3.83(d, J = 10.4 Hz, 1 H), 3.42(dd, J = 10.9, 6.2 Hz, 1H), 3.32(dd, J = 11.0, 5.5 Hz, 1H), 2.97 − 2.93(m, 1H), 2.82(dd, J = 14.8, 6.8 Hz, 1H), 2.42(t, J = 8.6 Hz, 1H), 2.00(dt, J = 12.7, 9.3 Hz, 1H), 1.92 − 1.85(m, 1H), 1.84 – 1.75(m, 1H), 1.67 − 1.57(m, 1H), 1.54 − 1.41(m, 5H), 1.38(s, 9H), 1.32(d, J = 13.3 Hz, 1H), 1.24 − 1.16(m, 1H), 1.09(d, J = 6.2 Hz, 1H), 1.00(s, 3H), 0.97(s, 3H). HRMS(ESI): m/z [M + H]+ calculated for C26H41NO8:
518.2730 ; found:518.2724 。 -
将化合物2(149 mg, 0.2 mmol)加入到25 ml单口瓶中,然后加入2 ml二氯甲烷和TFA(1.5 ml,19.5 mmol),室温下反应1.5 h。反应结束后减压蒸去溶剂,得122 mg黄色晶体3,收率79.8%。1H NMR(600 MHz, D2O)δ 4.99(d, J = 1.5 Hz, 1H), 4.16(d, J = 10.2 Hz, 1H), 4.03(d, J = 10.2 Hz, 1H), 3.57(t, J = 7.3 Hz, 1H), 3.48 – 3.45(m, 1H), 3.09 − 2.95(m, 3H), 2.55(t, J = 8.1 Hz, 1H), 2.05 − 1.95(m, 3H), 1.87 − 1.84(m, 1H), 1.74–1.69(m, 1H), 1.63 − 1.56(m, 1H), 1.56 − 1.47(m, 1H), 1.47 − 1.35(m, 3H), 1.27–1.25(m, 1H), 1.22–1.18(m, 1H), 1.00(s, 3H), 0.93(s, 3H)。
-
向10 ml单口瓶中分别加入化合物3(51 mg, 0.1 mmol)、2 ml二氯甲烷和三乙胺25.9 μl(20 mg, 0.2 mmol),冷却至0 oC,缓慢加入对甲苯磺酰异氰酸酯(19.7 mg, 0.1 mmol),缓慢升至室温反应过夜。反应结束后加入5 ml水,二氯甲烷萃取(5 ml × 3),合并有机相。有机相用水洗涤(10 ml),无水Na2SO4干燥,减压蒸去溶剂,柱层析纯化得30 mg白色固体(二氯甲烷∶甲醇=100∶5),收率25.9%。1H NMR(600 MHz, DMSO-d6)δ 10.59(s, 1H), 7.77(d, J = 8.3 Hz, 2H), 7.39(d, J = 8.0 Hz, 2H), 6.77(s, 1H), 6.57(s, 1H), 6.07(d, J = 2.2 Hz, 1H), 5.66(d, J = 10.9 Hz, 1H), 4.77(s, 1H), 4.34(d, J = 5.0 Hz, 1H), 4.05(d, J = 10.2 Hz, 1H), 3.82(d, J = 10.8 Hz, 1H), 3.41(dd, J = 11.0, 6.2 Hz, 1H), 3.29(dd, J = 10.8, 5.2 Hz, 1H), 2.98(dd, J = 13.3, 7.0 Hz, 2H), 2.73(s, 1H), 2.39(s, 3H), 2.36 – 2.32(m, 1H), 1.96(dd, J = 13.5, 7.9 Hz, 1H), 1.80(dd, J = 19.6, 10.1 Hz, 1H), 1.72(dd, J = 13.5, 6.5 Hz, 1H), 1.58(dd, J = 13.2, 6.2 Hz, 1H), 1.53 – 1.37(m, 4H), 1.33(d, J = 11.9 Hz, 2H), 1.18(dd, J = 15.2, 12.0 Hz, 1H), 1.06(d, J = 5.0 Hz, 1H), 0.99(s, 3H), 0.96(s, 3H).13C NMR(151 MHz, DMSO-d6)δ 224.71, 129.23, 128.93, 128.51, 127.12, 97.02, 73.66, 73.52, 71.92, 63.23, 61.67, 60.54, 52.97, 48.52, 45.88, 40.58, 38.83, 38.14, 36.92, 33.80, 32.97, 29.73, 26.34, 21.91, 21.38, 19.62, 18.71.HRMS(ESI): m/z [M + Na]+ calculated for C29H40N2O9S:
615.2353 ; found:615.2347 。 -
参考化合物ZM658的合成操作步骤,以苯磺酰异氰酸酯代替对甲苯磺酰异氰酸酯,得白色固体,收率17.3%。1H NMR(600 MHz, DMSO-d6) δ 10.66(s, 1H), 7.91 – 7.86(m, 2H), 7.66(t, J = 7.3 Hz, 1H), 7.59(t, J = 7.7 Hz, 2H), 6.77(s, 1H), 6.56(s, 1H), 6.06(d, J = 1.8 Hz, 1H), 5.66(d, J = 10.9 Hz, 1H), 4.77(s, 1H), 4.33(d, J = 5.0 Hz, 1H), 4.05(d, J = 10.2 Hz, 1H), 3.82(dd, J = 10.2, 1.3 Hz, 1H), 3.41(dd, J = 11.0, 6.2 Hz, 1H), 3.30 – 3.27(m, 1H), 2.98(dd, J = 13.2, 6.6 Hz, 2H), 2.74(dd, J = 15.1, 6.5 Hz, 1H), 2.35(t, J = 8.7 Hz, 1H), 1.95(td, J = 13.2, 7.6 Hz, 1H), 1.87 – 1.78(m, 1H), 1.74(td, J = 13.3, 7.4 Hz, 1H), 1.58(dd, J = 12.7, 6.2 Hz, 1H), 1.52 – 1.33(m, 5H), 1.30(dd, J = 10.1, 3.3 Hz, 1H), 1.18(td, J = 13.6, 3.2 Hz, 1H), 1.07(d, J = 6.1 Hz, 1H), 0.99(s, 3H), 0.96(s, 3H).13C NMR(151 MHz, DMSO-d6) δ 224.45, 152.19, 141.13, 133.38, 129.40, 127.51, 97.03, 73.62, 73.55, 71.93, 63.24, 61.65, 60.53, 52.94, 49.06, 48.43, 38.84, 38.18, 37.04, 33.80, 32.98, 29.74, 26.01, 21.92, 19.61, 18.70. HRMS(ESI): m/z [M + Na]+ calculated for C28H38N2O9S:
601.2196 ; found:601.2190 。 -
选用人单核细胞白血病细胞(THP-1,普诺赛)作为试验细胞株。所有目标化合物用药用级DMSO配置成对应浓度的药液,阳性药物以同样的条件配成对照品溶液。采用CCK-8法测化合物细胞毒性。将THP-1细胞置于96孔板中,用100 ng/ml PMA刺激48 h。取上清液,用含有Ori或不同浓度目标化合物的新鲜培养基替换。然后用Ori或不同浓度目标化合物处理细胞24 h。用含有CCK8(10 μl)的新鲜培养基替换培养上清液,并在37 ◦C下继续培养3 h。用酶标仪在450 nm处记录吸光度(A)值,根据A值计算细胞活力。
$$ \begin{split}\mathrm{Cell}\;\mathrm{viability}(\text{%})=&\left[({{A}}_{\mathrm{test}}-{{A}}_{\mathrm{blank}})/({{A}}_{\mathrm{control}}-{{A}}_{\mathrm{blank}})\right]\times \\&100\text{%}\end{split} $$ THP-1细胞在完全培养基(含10%胎牛血清、1%青链霉素合剂和0.05 mmol β-巯基乙醇的RPMI1640培养基)、含有5%二氧化碳和95%空气的37 ◦C的无菌环境中培养。吸出培养瓶中的含THP-1细胞的培养基,1 000 r/min离心3 min,用完全培养基重悬细胞,调整细胞浓度为5105 个/ml,接种于24孔板中,每孔细胞培养基混合液1 ml。加入浓度为100 ng/ml的PMA,刺激THP-1细胞48 h。将PMA分化的THP-1细胞用磷酸盐缓冲盐水洗涤,然后在基础培养基中用100 μg/ml的LPS处理3 h。去除培养基,向24孔板中分别加入各浓度的化合物培养基,混匀,孵箱孵育处理24 h,ATP(5 mmol/L)刺激细胞 1 h。收集细胞培养上清液,12 000 r/min离心5 min,去除死细胞,使用ELISA法分析细胞上清液中的1L-1β抑制率。
-
光催化反应因其具有高清洁性和高效率等优点,在药物研究领域逐渐得到应用。本课题组以冬凌草甲素(1)为起始原料,在磷酸氢二钾和Ir[dF(CF3)ppy]2(dtbbpy)PF6催化和LED光照射下,与Boc-甘氨酸发生加成反应,以较高收率得到C-16位带有氨基侧链的关键中间体(2)。然后,关键中间体(2)脱去Boc保护基得到氨基中间体(3)。最后,以三乙胺为缚酸剂,与取代异氰酸酯反应,合成得到2个目标产物(图2)。整条合成路线共有3步反应,反应条件温和,后处理和分离纯化较简单。
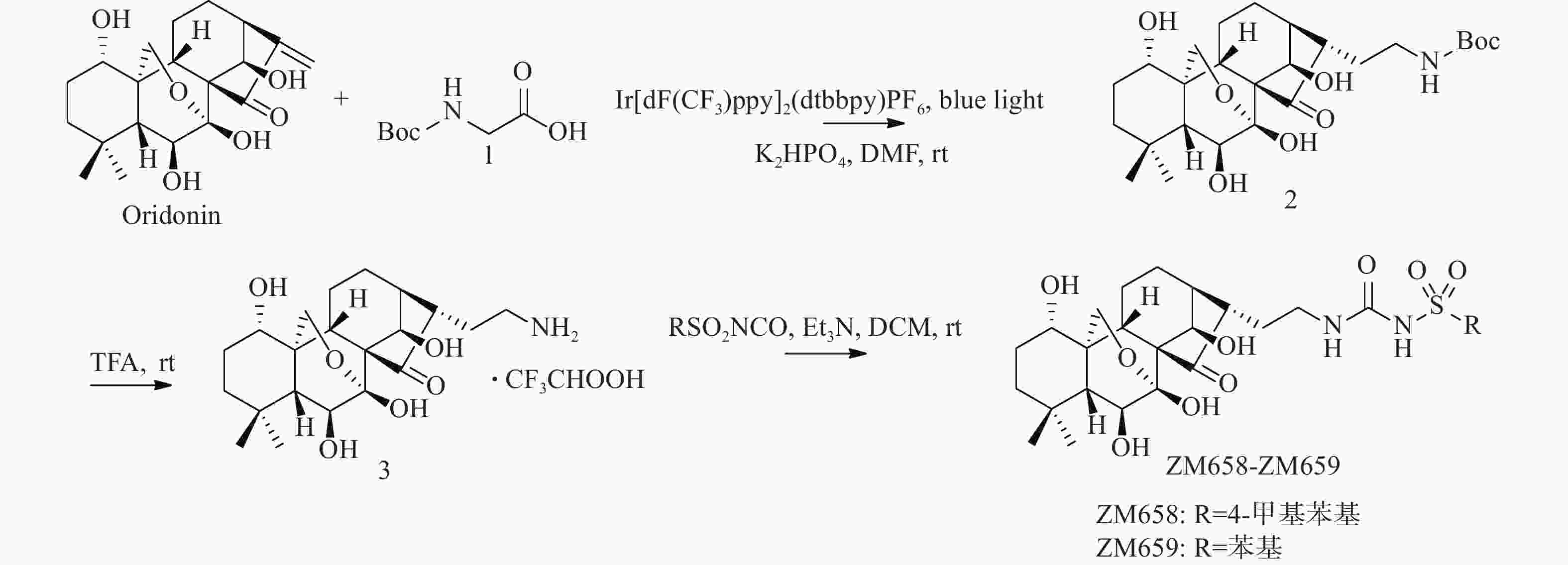
图 2 冬凌草甲素磺酰脲衍生物的合成路线
-
本课题组使用ATP作为NLRP3炎症小体激活剂,采用肉豆蔻酸乙酯刺激THP-1细胞分化的巨噬细胞模型进行体外抗炎活性测试[17]。采用ELISA法检测IL-1β分泌水平,用以评估目标化合物对NLRP3炎症小体的抑制作用,并以先导化合物冬凌草甲素为阳性对照。
首先,本课题组采用MTT法测试了新化合物的毒性,测试结果如表1。从表中可以看出,将冬凌草甲素的迈克尔受体片段α , β -不饱和酮的双键替换成苯磺酰脲后,化合物的细胞毒性均得到了大幅下降,CC50值均大于100 μmol。抗炎活性研究结果表明,将冬凌草甲素的迈克尔受体片段中双键去除后,几乎丧失了抗炎活性,如中间体2和3无论是在20 μmol/L浓度下还是50 μmol/L浓度下,IL-1β的抑制率均很低。但将磺酰脲片段引入后显示出优秀的抗炎活性,其中,化合物ZM658在20 μmol/L浓度时,IL-1β的抑制率为69.3%,优于冬凌草甲素;化合物ZM659对IL-1β的抑制率也达59.7%,稍低于冬凌草甲素。然而,在高浓度(50 μmol/L)时,冬凌草甲素磺酰脲衍生物的抗炎活性均低于先导物冬凌草甲素。
表 1 冬凌草甲素磺酰脲衍生物的抗炎活性
化合物 IL-1β抑制率(%) CC50(μmol/L) 20 μmol/L 50 μmol/L ZM658 69.3 75.7 >100 ZM659 59.7 60.8 >100 2 9.8 22.8 >100 3 −8 4.0 >100 冬凌草甲素 63.0 90.1 19.8 -
基于光催化反应和骨架跃迁药物设计策略,以磺酰脲片段代替冬凌草甲素的关键药效团——迈克尔受体片段,设计合成出2个冬凌草甲素磺酰脲衍生物。细胞毒活性测试表明,新化合物的毒性较小,低于先导物冬凌草甲素。抗炎活性研究发现,冬凌草甲素磺酰脲衍生物均显示出优秀的抗炎活性,化合物ZM658在20 μmol/L浓度时,对IL-1β的抑制率优于冬凌草甲素,并且随着浓度的提高也呈现出一定的量效关系。本研究结果初步表明,冬凌草甲素的迈克尔受体片段并非是其作为NLRP3炎症小体抑制剂的必需药效团,这为以冬凌草甲素为骨架的新型NLRP3炎症小体抑制剂的进一步研究提供了新的优化设计思路。
Synthesis and anti-inflammatory activity of oridonin sulfonylurea derivatives
-
摘要:
目的 研究迈克尔结构片段对冬凌草甲素抗炎活性的影响作用。 方法 基于光催化反应和骨架跃迁策略,设计合成出2个冬凌草甲素磺酰脲类化合物;以IL-1β抑制率为指标,进行新化合物的抗炎活性评价。 结果 目标化合物均显示出较好的抗炎活性,在20 μmol/L浓度下,化合物 ZM658 和 ZM659 对人单核细胞白血病THP-1细胞中炎症因子IL-1β抑制率分别为69.3%和59.7%,并且显示出一定的量效关系和低毒性。 结论 以磺酰脲代替冬凌草甲素的迈克尔受体结构片段后可保持化合物的抗炎活性。 Abstract:Objective To study anti-inflammatory activities of oridonin derivatives without Michael fragment. Methods Two oridonin sulfonylureas were designed and synthesized by a photocatalysis reaction and a scaffold hopping strategy. The inhibitory rate of IL-1β was selected for anti-inflammatory activity evaluation. Results Both compound ZM658 and ZM659 revealed potent anti-inflammatory activities with the values of 69.3% and 59.7% in THP-1 cells, respectively. Moreover, two compounds also showed dose-dependent and low cytotoxicity. Conclusion The result indicated that Michael receptor fragment of oridonin could be substituted with sulfonylurea group. -
Key words:
- oridonin /
- photocatalysis reaction /
- scaffold hopping /
- synthesis /
- anti-inflammatory activity
-
表 1 冬凌草甲素磺酰脲衍生物的抗炎活性
化合物 IL-1β抑制率(%) CC50(μmol/L) 20 μmol/L 50 μmol/L ZM658 69.3 75.7 >100 ZM659 59.7 60.8 >100 2 9.8 22.8 >100 3 −8 4.0 >100 冬凌草甲素 63.0 90.1 19.8  下载: 导出CSV
下载: 导出CSV
-
[1] FU J N, WU H. Structural mechanisms of NLRP3 inflammasome assembly and activation[J]. Annu Rev Immunol, 2023, 41:301-316. [2] 邓波, 霍亚南. NLRP3炎症小体与代谢性疾病关系研究进展[J]. 中国当代医药, 2023, 30(17):24-27. doi: 10.3969/j.issn.1674-4721.2023.17.007 [3] MANGAN M S J, OLHAVA E J, ROUSH W R, et al. Targeting the NLRP3 inflammasome in inflammatory diseases[J]. Nat Rev Drug Discov, 2018, 17(8):588-606. [4] KELLEY N, JELTEMA D, DUAN Y H, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation[J]. Int J Mol Sci, 2019, 20(13):3328. [5] BLEVINS H M, XU Y M, BIBY S, et al. The NLRP3 inflammasome pathway: a review of mechanisms and inhibitors for the treatment of inflammatory diseases[J]. Front Aging Neurosci, 2022, 14:879021. [6] MA Q. Pharmacological inhibition of the NLRP3 inflammasome: structure, molecular activation, and inhibitor-NLRP3 interaction[J]. Pharmacol Rev, 2023, 75(3):487-520. doi: 10.1124/pharmrev.122.000629 [7] ZAHID A, LI B F, KOMBE A J K, et al. Pharmacological inhibitors of the NLRP3 inflammasome[J]. Front Immunol, 2019, 10:2538. [8] YANG Y, WANG H N, KOUADIR M, et al. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors[J]. Cell Death Dis, 2019, 10(2):128. doi: 10.1038/s41419-019-1413-8 [9] DE BENEDETTI F, GATTORNO M, ANTON J, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes[J]. N Engl J Med, 2018, 378(20):1908-1919. doi: 10.1056/NEJMoa1706314 [10] LI H, GUAN Y L, LIANG B, et al. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome[J]. Eur J Pharmacol, 2022, 928:175091. [11] BAKHSHI S, SHAMSI S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: latest evidence and therapeutic outcomes[J]. Int Immunopharmacol, 2022, 106:108595. doi: 10.1016/j.intimp.2022.108595 [12] DAI Z, CHEN X Y, AN L Y, et al. Development of novel tetrahydroquinoline inhibitors of NLRP3 inflammasome for potential treatment of DSS-induced mouse colitis[J]. J Med Chem, 2021, 64(1):871-889. doi: 10.1021/acs.jmedchem.0c01924 [13] WANG L, ZHAO X Q, DING J W, et al. Oridonin attenuates the progression of atherosclerosis by inhibiting NLRP3 and activating Nrf2 in apolipoprotein E-deficient mice[J]. Inflammopharmacology, 2023, 31(4):1993-2005. doi: 10.1007/s10787-023-01161-9 [14] HE H B, JIANG H, CHEN Y, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity[J]. Nat Commun, 2018, 9(1):2550. [15] 徐博, 王茂楠. 冬凌草甲素对DSS诱导的小鼠溃疡性结肠炎的保护作用[J]. 吉林医学, 2023, 44(5):1148-1151. doi: 10.3969/j.issn.1004-0412.2023.05.002 [16] BELLOTTI P, ROGGE T, PAULUS F, et al. Visible-light photocatalyzed peri-(3 + 2)cycloadditions of quinolines[J]. J Am Chem Soc, 2022, 144(34):15662-15671. [17] LI M, WANG C H, YE S, et al. Discovery of novel oridonin sulfamide derivatives as potent NLRP3 inhibitors by a visible-light photocatalysis-enabled peripheral editing[J]. Bioorg Med Chem Lett, 2024, 99:129621. -
 点击查看大图
点击查看大图
图(2) / 表(1)
计量
- 文章访问数: 231
- HTML全文浏览量: 145
- PDF下载量: 0
- 被引次数: 0

