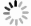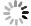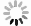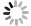


-
冠状病毒(coronavirus, CoV)是一类具有单股正链RNA基因组的包膜病毒,基因组长度为26~32 kb[1]。这类病毒广泛分布于哺乳动物和鸟类中,能够引起从轻微的呼吸道感染到严重的呼吸道综合征等多种疾病[2-3]。2019年底出现的SARS-CoV-2,引发了全球冠状病毒病(COVID-19)大流行,造成了巨大的公共卫生危机[4]。SARS-CoV-2是人类发现的第7种冠状病毒,属于包膜内线性单链正向RNA病毒,包含14个开放阅读框,其中,ORF1a和ORF1b编码的多聚蛋白在病毒复制和转录过程中起关键作用[5-6]。这些多聚蛋白在主蛋白酶(main protease, Mpro,也称为3C-like protease, 3CLpro)和木瓜样蛋白酶(papain-like proteinase protein, PLpro)的作用下被切割成多个非结构蛋白,进一步组装成病毒复制转录复合体,从而启动病毒RNA的复制和转录[7-9]。Mpro在病毒复制感染中扮演着至关重要的角色,其底物识别位点在所有冠状病毒中高度保守,且与人类蛋白酶没有显著的同源性,使其成为极具潜力的抗冠状病毒药物治疗靶点[10]。
近年来,针对SARS-CoV-2 Mpro的抑制剂研究取得了显著进展,已有多个药物获得临床批准用于SARS-CoV-2病毒感染的治疗,如辉瑞公司的奈玛特韦(Nirmatrelvir)、先声药业的先诺欣以及众生睿创的乐睿灵。此外,还有多个Mpro抑制剂处于新药研发阶段。然而,现有抑制剂主要通过与Mpro活性位点的可逆或不可逆结合来发挥功能,存在一些局限性[11-13]:①传统抑制剂需要较高浓度才能有效抑制病毒复制,可能增加药物毒性和副作用的风险;②病毒突变会显著降低药物结合亲和力,导致耐药性,从而降低抑制剂的疗效。例如,SARS-CoV-2 Mpro的E166V和E166A突变已被证实对奈玛特韦表现出显著的耐药性[14]。
为了克服这些问题,蛋白降解靶向嵌合体(proteolysis targeting chimera, PROTAC)作为一种新兴的药物研发技术,为抗病毒药物的研发提供了新的思路[15-17]。与传统小分子抑制剂依靠占据靶蛋白活性位点来阻断其功能不同,PROTAC分子一端与靶蛋白结合,另一端结合E3泛素连接酶形成靶蛋白-PROTAC-E3三元复合物,通过招募E3连接酶,诱导靶蛋白的泛素化和随后的蛋白酶体降解。这种事件驱动的作用机制不仅提高了药物的选择性和作用效力,还能有效克服因靶蛋白突变导致的耐药性问题[18-19]。在抗病毒药物研究中,PROTAC技术已应用于多种病毒蛋白的降解,如丙型肝炎病毒NS3蛋白和流感病毒血凝素蛋白。针对SARS-CoV-2的Mpro蛋白,也有相关研究报道。Alugubelli等[20-21]报道的Mpro PROTAC MPD2在细胞中表现出显著的Mpro蛋白降解活性和抗病毒效果。Sang等[22]也报道了一种基于CRBN配体的PROTAC分子HP211206,能够有效降解SARS-CoV-2 Mpro及其耐药突变体。
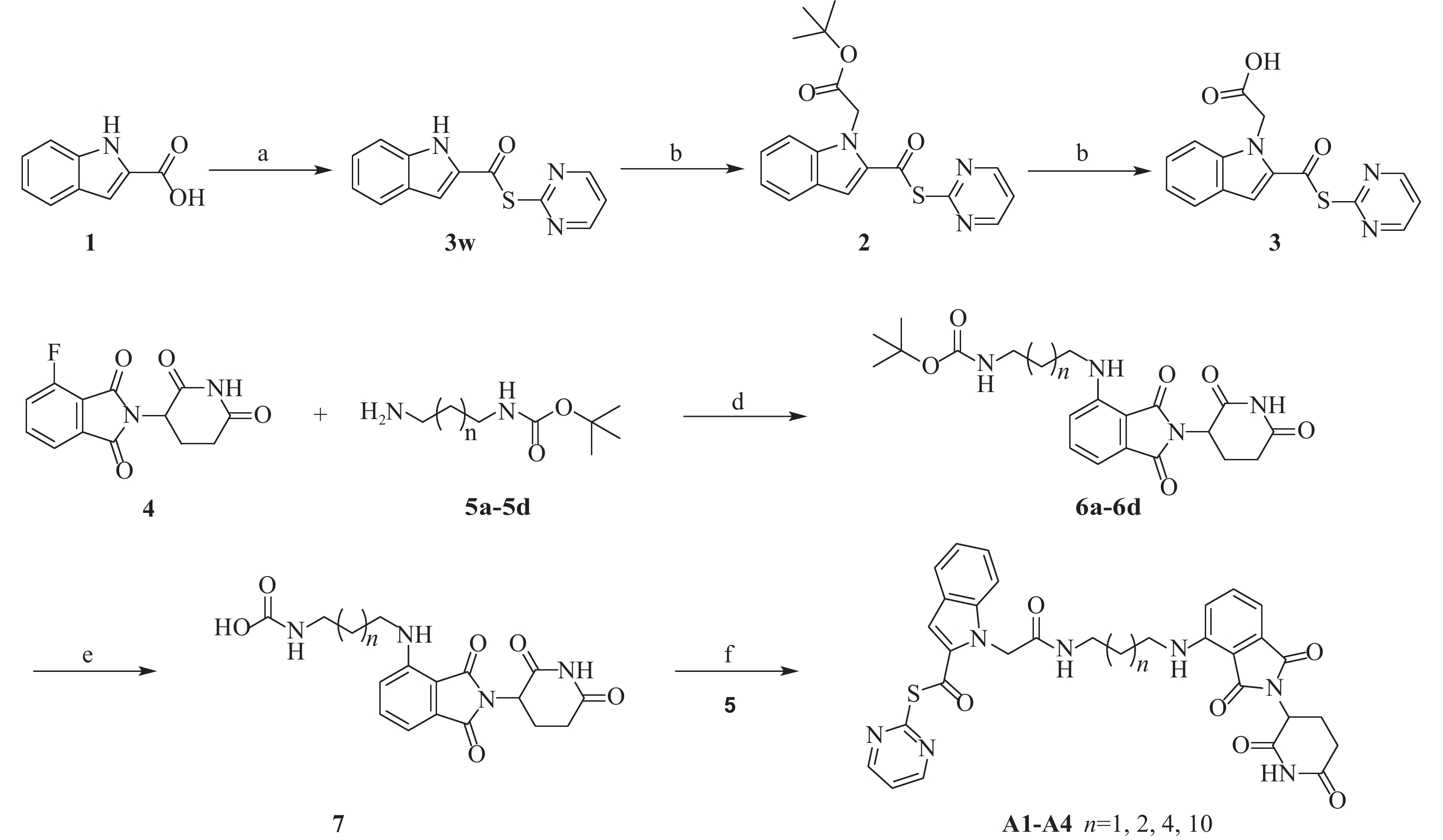
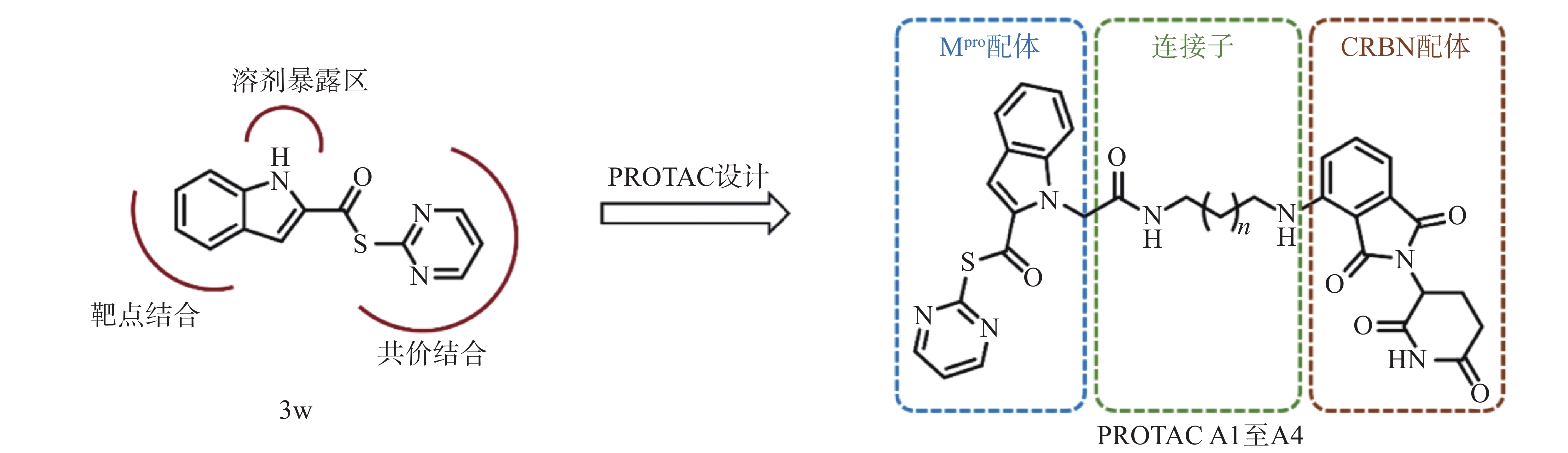
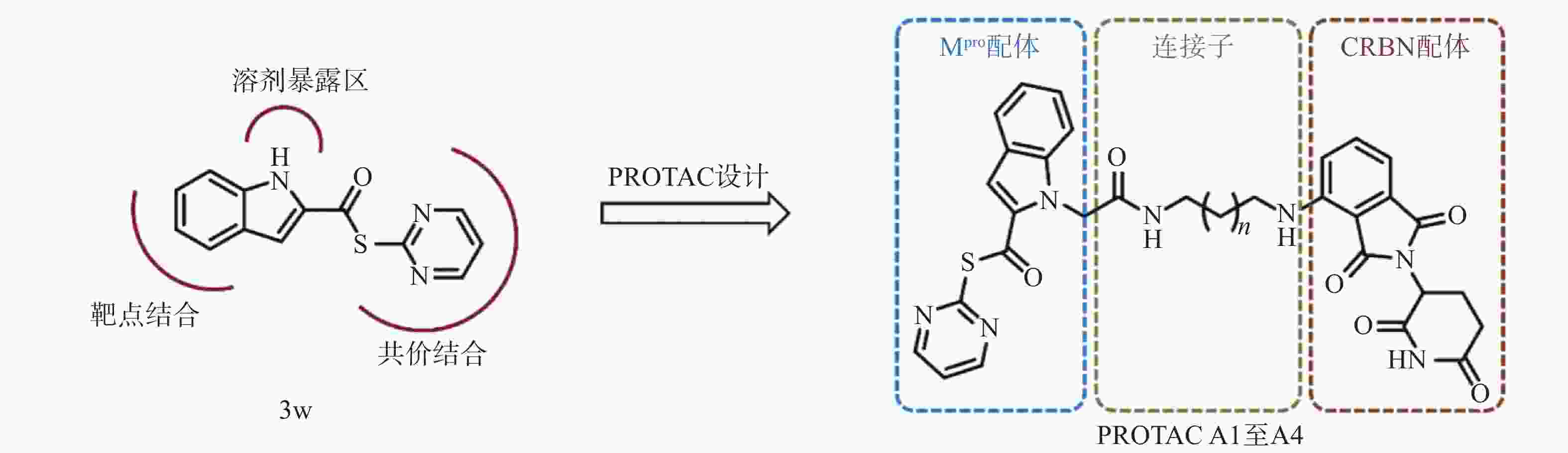
然而,当前针对Mpro的PROTAC降解剂大多基于复杂的拟肽类抑制剂,其合成的难度和成本较高,为后续研发和生产带来一定挑战[23]。为了探索更简单、高效的小分子抗新冠病毒PROTAC降解剂,我们设计并合成一类基于简单小分子抑制剂3w的Mpro PROTAC降解剂[24](图1)。化合物3w是一种硫酯类小分子Mpro共价抑制剂,与传统Mpro抑制剂相比,具有分子量小、结构简单、易于合成的优点。本研究旨在验证基于简单小分子配体的PROTAC策略在抗SARS-CoV-2药物研究中的可行性,为未来抗病毒药物的设计提供新的先导化合物。
-
化学原料均为分析纯或化学纯级别,购自探索平台、毕得等试剂公司;Rabbit anti-SARS-CoV-2 3CLpro antibody(ABclonal, A20831);Rabbit anti-β-Tubulin antibody(ABclonal,A12289);Goat anti-rabbit IgG H&L(Alexa Fluor® 680)(Abcam, ab175773);Bruker AVANCE600(Bruker Company, Germany)核磁共振仪,TMS作为内标,化学位移与偶合常数分别用ppm和Hz表示;Agilent
6538 UHD Accurate-Mass Q-TOF LC/MS高分辨质谱(HRMS)仪;Bioteck Synergy2多功能酶标仪;Biorad ChemiDoc成像仪。 -
合成路线见图2。
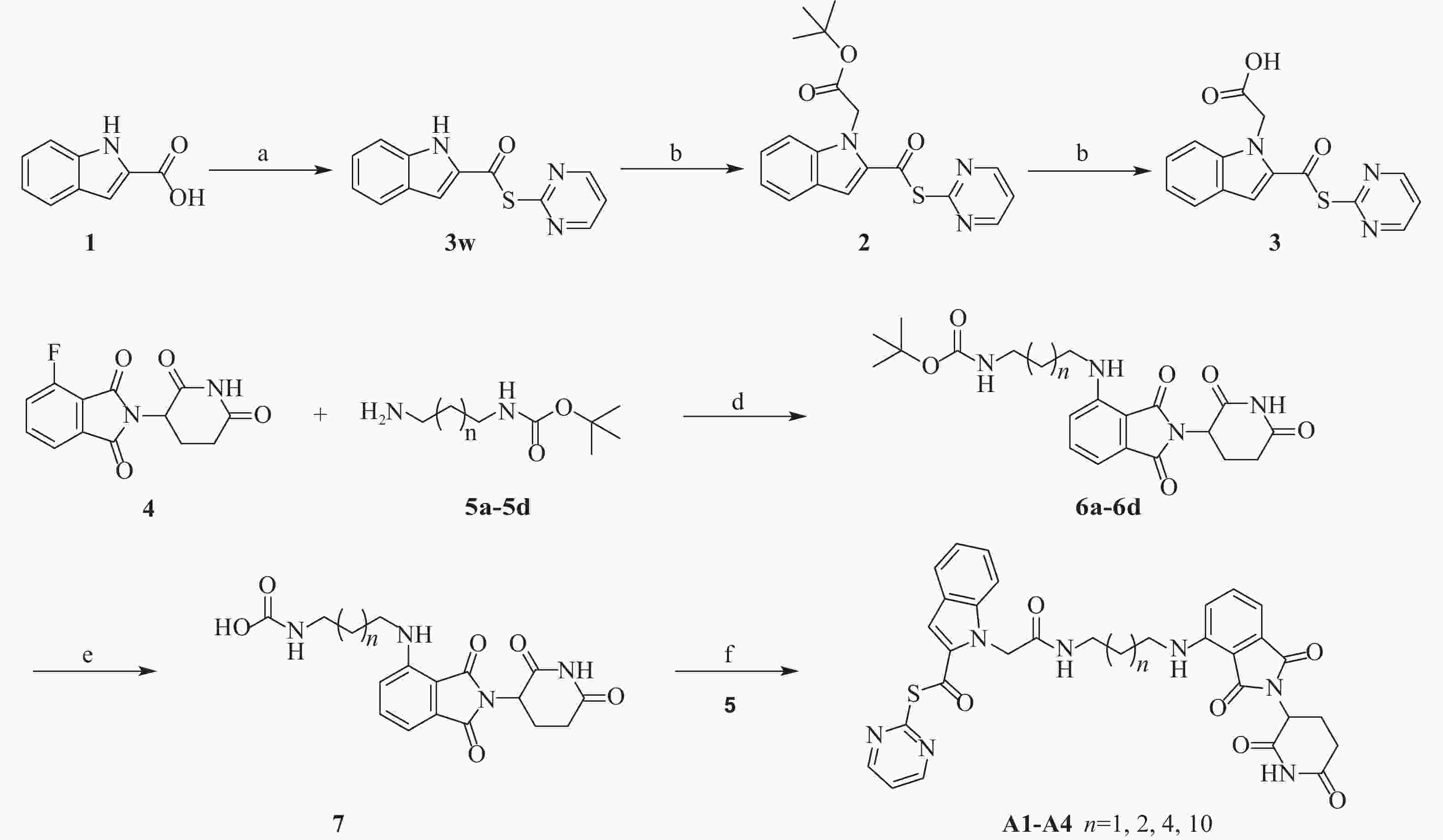
-
将1H-吲哚-2-羧酸(3.0 g, 18 mmol)溶于无水二氯甲烷(DCM, 30 ml),氮气保护下依次加入三氯氧磷(4.3 g, 28 mmol)和吡啶(2.2 g, 28 mmol)。室温搅拌30 min后,加入嘧啶-2-硫醇(0.70 g, 18 mmol)及吡啶(2.4 g, 30 mmol),室温反应8 h。反应液用饱和柠檬酸钠水溶液(50 ml)淬灭,DCM(20 ml× 3)萃取。合并有机相,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩得粗产物,经硅胶柱层析纯化(石油醚/乙酸乙酯= 60:40)得白色固体(3w)1.86 g,收率61%。1H NMR(600 MHz, DMSO-d6)δ 12.14(s, 1H), 8.93(d, J = 4.9 Hz, 2H), 7.75(d, J = 8.1 Hz, 1H), 7.59(t, J = 4.9 Hz, 1H), 7.51(s, 1H), 7.48(d, J = 8.4 Hz, 1H), 7.34(t, J = 7.7 Hz, 1H), 7.14(t, J = 7.6 Hz, 1H)。 13C NMR(101 MHz, DMSO-d6)δ 178.7, 163.7, 159.1, 138.3, 132.9, 126.6, 126.3, 122.8, 120.9, 120.9, 112.9, 109.6. ESI-HRMS m/z Calcd. For C13H9N3OS [M+H]+
256.0545 , Found 256.0539。 -
将化合物3w(0.22 g,0.85 mmol)与2-溴乙酸叔丁酯(0.25 g,1.28 mmol)溶于丙酮(20 ml),随后加入Cs2CO3(0.83 g,2.55 mmol),室温搅拌6 h。反应液减压抽滤得白色固体,经硅胶柱层析纯化(石油醚/乙酸乙酯= 65∶35),得淡黄色固体(2)0.18 g,收率66%。1H NMR(600 MHz, DMSO-d6)δ 8.92(d, J = 4.9 Hz, 2H), 7.97(d, J = 8.4 Hz, 1H), 7.84(d, J = 8.0 Hz, 1H), 7.67(s,1H), 7.53(t, J = 4.9 Hz, 1H), 7.45(t, J = 7.9 Hz, 1H), 7.31(t, J = 7.5 Hz, 1H), 3.82(s, 2H), 1.40(s, 9H)。13C NMR(101 MHz, DMSO-d6)δ 184.2, 172.5, 159.5, 156.9, 138.7, 136.4, 132.1, 129.2, 127.4, 123.4, 123.3, 119.7, 113.5, 80.0, 35.2, 27.6。
-
将上述化合物2(0.18 g, 0.49 mmol)溶于DCM(20 ml),加入三氟乙酸(TFA, 4 ml),室温搅拌6 h,待反应完全后旋干溶剂和TFA,得淡黄色固体(3)0.17 g,收率95%。
-
将2-(2,6-二氧代哌啶-3-基)-4-氟异吲哚啉-1,3-二酮(0.27 g, 0.96 mmol)与N-叔丁氧羰基-1,3-丙二胺(0.25 g, 1.43 mmol)溶于N-甲基吡咯烷酮溶液中,逐滴加入N,N-二异丙基乙胺(DIPEA)(1.2 g, 9.6 mmol)。将反应混合物在90 ℃下加热搅拌5 h。待反应结束后,反应液用饱和氯化钠水溶液(20 ml)溶解,然后用乙酸乙酯萃取(10 ml× 3),合并有机相,无水硫酸钠干燥,减压浓缩。粗产物经硅胶柱层析纯化(石油醚/乙酸乙酯=55∶45),得黄色固体(6a)0.26 g,收率65%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 7.57(t, J = 7.8 Hz, 1H), 7.08(d, J = 8.6 Hz, 1H), 7.02(d, J = 7.0 Hz, 1H), 6.92(s, 1H), 6.66(s, 1H), 5.05(dd, J = 12.9, 5.4 Hz, 1H), 3.30(t, J = 7.2 Hz, 2H), 3.03 - 2.98(m, 2H), 2.61 - 2.52(m, 2H), 1.69 - 1.61(m, 2H), 1.38(s, 9H), 1.29 - 1.23(m, 2H)。
-
中间体6b的合成步骤参照中间体6a,得到黄色固体(6b)0.19 g,收率58%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 7.56(t, J = 8.4 Hz, 1H), 7.09(d, J = 9.0 Hz, 1H), 7.01(d, J = 9.0 Hz, 1H), 6.81(s, 1H), 6.53(s, 1H), 5.03(dd, J = 11.4, 10.8 Hz, 1H), 3.28(t, J = 9.0 Hz, 2H), 2.94 - 2.92(m, 2H), 2.69 - 2.67(m, 2H), 1.55 - 1.52(m, 2H), 1.49 - 1.38(m, 2H), 1.35(s, 9H), 1.33 - 1.08(m, 2H)。
-
中间体6c的合成步骤参照中间体6a,得到黄色固体(6c)0.22 g,收率72%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 7.58(t, J = 7.2 Hz, 1H), 7.09(d, J = 8.7 Hz, 1H), 7.02(d, J = 7.2 Hz, 1H), 6.74(s, 1H), 6.52(s, 1H), 5.05(dd, J = 7.5, 2.8 Hz, 1H), 3.30(t, J = 3.0 Hz, 2H), 2.91 - 2.84(m, 2H), 2.61 - 2.51(m, 2H), 1.57 - 1.54 (m, 2H), 1.36(s, 9H), 1.35 - 1.03(m, 12H)。
-
中间体6d的合成步骤参照中间体6a,得到黄色固体(6d)0.21 g,收率69%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 7.57(t, J = 7.2 Hz 1H), 7.08(d, J = 8.6 Hz, 1H), 7.01(d, J = 7.0 Hz, 1H), 6.73(s, 1H), 6.52(s, 1H), 5.04(dd, J = 12.9, 5.4 Hz, 1H), 3.28(t, J = 6.6 Hz, 2H), 2.88 - 2.87(m, 2H), 2.61 - 2.51(m, 2H), 1.60 - 1.49(m, 2H), 1.36(s, 9H), 1.30 - 1.21(m, 20H)。
-
将6a溶于DCM(20 ml),随后加入TFA(4 ml),在室温下搅拌6 h。旋蒸除去溶剂和TFA后,向残留物中加入化合物3(0.19 g, 0.62 mmol),并将其溶于DCM(15 ml)中。在室温下搅拌,缓慢滴加DIPEA,直至不产生白烟。称取1-乙基-3-(3-二甲氨基丙基)碳二亚胺盐酸盐(EDCI, 0.13 g, 0.68 mmol),1-羟基苯并三唑(HOBt, 0.092 g, 0.68 mmol)溶于DCM中,随后将混合液滴加到反应液中,最后滴加DIPEA(0.24 g, 1.86 mmol),继续在室温下搅拌反应10 h。反应结束后,反应液用饱和碳酸氢钠溶液(50 ml)淬灭,DCM(20 ml× 3)萃取。合并有机相后,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,减压浓缩除去溶剂。残余物经硅胶柱层析纯化(二氯甲烷/甲醇= 95:5),得淡黄色固体(A1)0.11 g,收率33%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 8.91(d, J = 4.9 Hz, 2H), 8.27(t, J = 5.7 Hz, 1H), 7.96(d, J = 8.5 Hz, 1H), 7.83(d, J = 7.9 Hz, 1H), 7.66(s, 1H), 7.53 - 7.48(m, 1H), 7.49(t, J = 4.8 Hz, 1H), 7.44(t, J = 7.7 Hz, 1H) , 7.31(t, J = 7.5 Hz, 1H), 7.06(d, J = 8.6 Hz, 1H), 6.99(d, J = 7.0 Hz, 1H), 6.67(t, J = 6.2 Hz, 1H), 5.04(dd, J = 12.8, 5.5 Hz, 1H), 3.76(s, 2H), 3.31 - 3.28(m, 2H), 3.16(q, J = 6.3, 6.3, 6.3 Hz, 2H), 2.63 - 2.51(m, 2H), 1.70 - 1.65(m, 2H), 1.36 - 1.23(m, 2H)。13C NMR(101 MHz, DMSO-d6)δ 182.8, 172.8, 170.1, 168.8, 167.3, 166.8, 158.9, 156.3, 146.2, 138.4, 136.2, 135.1, 132.2, 127.1, 126.6, 123.0, 119.3, 117.1, 114.2, 112.9, 110.4, 109.2, 48.5, 36.4, 32.8, 30.9, 28.9, 28.6, 22.2。ESI-HRMS m/z Calcd. For C31H27N7O6S [M+H]+
626.1822 , Found626.1816 。 -
目标化合物A2合成步骤参照化合物A1,得淡黄色固体(A2)0.096 g,收率31%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 8.92(d, J = 4.9 Hz, 2H), 8.19(t, J = 5.7 Hz, 1H), 7.96(d, J = 8.5 Hz, 1H), 7.82(d, J = 8.0 Hz, 1H), 7.64(s, 1H), 7.57 - 7.53(m, 1H), 7.52(t, J = 5.2 Hz, 1H), 7.44(t, J = 7.7 Hz, 1H), 7.30(t, J = 7.5 Hz, 1H), 7.08(d, J = 8.5 Hz, 1H), 6.99(d, J = 7.0 Hz, 1H), 6.54(t, J = 5.8 Hz, 1H), 5.04(dd, J = 12.5, 5.4 Hz, 1H), 3.74(s, 2H), 3.31 - 3.25(m, 2H), 3.11(q, J = 6.5, 6.5, 6.5 Hz, 2H), 2.63 - 2.52(m, 2H), 1.75 - 1.56(m, 2H), 1.62 - 1.54(m, 2H), 1.50 - 1.45(m, 2H)。13C NMR(101 MHz, DMSO-d6)δ 182.7, 172.8, 170.1, 168.9, 167.3, 166.5, 158.9, 158.5, 146.4, 138.4, 136.2, 135.1, 132.2, 127.1, 126.6, 122.9, 119.3, 117.2, 114.1, 112.9, 110.4, 109.0, 48.5, 41.5, 38.6, 32.7, 30.9, 26.1, 22.1。ESI-HRMS m/z Calcd. For C32H29N7O6S [M+H]+
640.1978 , Found640.1973 。 -
目标化合物A3合成步骤参照化合物A1,得淡黄色固体(A3)0.12 g,收率39%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 8.93(d, J = 4.9 Hz, 2H), 8.12(t, J = 5.7 Hz, 1H), 7.96(d, J = 8.5 Hz, 1H), 7.83(d, J = 8.0 Hz, 1H), 7.65(s, 1H), 7.58 - 7.55(m, 1H), 7.53(t, J = 4.9 Hz, 1H), 7.44(t, J = 7.8 Hz, 1H), 7.30(t, J = 7.5 Hz, 1H), 7.06(d, J = 8.6 Hz, 1H), 7.01(d, J = 7.0 Hz, 1H), 6.50(t, J = 6.0 Hz, 1H), 5.05(dd, J = 12.8, 5.5 Hz, 1H), 3.73(s, 2H), 3.28 - 3.23(m, 2H), 3.05(q, J = 6.6, 6.6, 6.6 Hz, 2H), 2.64 - 2.51(m, 2H), 1.56 - 1.51(m, 2H), 1.39 - 1.37(m, 2H), 1.33 - 1.23(m, 10H)。13C NMR(101 MHz, DMSO-d6)δ 182.7, 172.8, 170.1, 168.9, 167.3, 166.3, 158.9, 156.3, 146.4, 138.4, 136.2, 135.1, 132.2, 127.1, 126.6, 122.9, 119.3, 117.1, 114.2, 112.9, 110.3, 108.9, 48.5, 41.8, 32.7, 30.9, 30.1, 28.6, 26.2, 22.1, 17.2。ESI-HRMS m/z Calcd. For C36H37N7O6S [M+H]+ 696.2604, Found 696.2599。
-
目标化合物A4合成步骤参照化合物A1,得淡黄色固体(A4)0.14 g,收率42%。1H NMR(600 MHz, DMSO-d6)δ 11.09(s, 1H), 8.92(d, J = 4.9 Hz, 2H), 8.11(t, J = 5.6 Hz, 1H), 7.96(d, J = 8.5 Hz, 1H), 7.82(d, J = 8.0 Hz, 1H), 7.64(s, 1H), 7.58 - 7.55(m, 1H), 7.52(t, J = 4.8 Hz, 1H), 7.43(t, J = 7.8 Hz, 1H), 7.30(t, J = 7.5 Hz, 1H), 7.07(d, J = 8.6 Hz, 1H), 7.01(d, J = 7.1 Hz, 1H), 6.50(t, J = 5.8 Hz, 1H), 5.05(dd, J = 12.9, 5.4 Hz, 1H), 3.73(s, 2H), 3.27 - 3.24(m, 2H), 3.04(q, J = 6.7, 6.6, 6.6 Hz, 2H), 2.62 - 2.52(m, 2H), 1.56 - 1.51(m, 2H), 1.38 - 1.36(m, 2H), 1.32 - 1.25(m, 18H)。 13C NMR(101 MHz, DMSO-d6)δ 182.7, 172.8, 170.0, 168.9, 167.3, 166.3, 158.9, 158.5, 146.4, 138.4, 136.2, 135.1, 132.2, 127.1, 126.6, 122.9, 119.2, 117.1, 114.1, 112.9, 110.3, 109.0, 64.9, 48.5, 41.8, 32.7, 30.9, 28.9, 26.3, 22.1, 15.1。ESI-HRMS m/z Calcd. For C40H45N7O6S [M+H]+ 752.3230, Found 752.3230。
-
构建携带Mpro基因的质粒,并在其中插入编码荧光蛋白的基因序列,然后进行质粒抽提以获得高纯度的重组质粒。使用HG TransgeneTM Reagent转染试剂将构建好的质粒及慢病毒载体共转染进HEK-293T细胞。转染10~12 h后加Enhancing buffer,8 h后更换新鲜培养基,继续培养48 h,收集富含慢病毒颗粒的细胞上清液,对其浓缩后得到高滴度的慢病毒浓缩液。将HEK-293T细胞以2 × 105个细胞/孔密度接种于12孔板中,用800 μl完全培养基按感染复数(MOI)= 10稀释慢病毒原液,感染16 h后换成完全培养基,继续培养72 h,使细胞充分表达目标蛋白,最终获得稳定表达Mpro的HEK-293T细胞株(Mpro-HEK-293T)。通过蛋白印迹法(Western Blot)检测Mpro蛋白的表达水平,验证基因过表达效果。
-
将Mpro-HEK-293T细胞以4 × 105个/孔的密度接种于6孔板中,置于37 ℃、5% CO2的培养箱培养24 h,使细胞贴壁生长。取对数生长期的Mpro-HEK-293T细胞接种至6孔板。设置含不同药物浓度的完全培养基作为实验组,加入适量DMSO的完全培养基作为阴性对照组,只有完全培养基的作为空白对照组。各组细胞在相同条件下孵育24 h后提取细胞总蛋白。采用Western Blot检测细胞中Mpro的降解情况。首先,使用以10%十二烷基硫酸钠-聚丙烯酰胺凝胶电泳分离蛋白样品。随后,在恒压25 V条件下转膜8 min,将蛋白转移至聚偏二氟乙烯膜(PVDF)上。转膜后,将PVDF膜置于快速封闭液中,室温孵育封闭30 min。封闭结束后,用TBST缓冲液洗涤膜3次,每次5 min,然后与Mpro兔单克隆抗体(稀释比例1∶1 000)在4 ℃条件下孵育过夜。再次用TBST洗涤膜3次,每次5 min。随后与化学荧光兔二抗在室温下孵育2 h。最后,将膜置于BioRad ChemiDoc成像仪中进行化学发光信号检测并曝光成像。
-
为了评估目标化合物对Mpro在细胞中的降解活性,我们构建了稳定表达SARS-CoV-2 Mpro的HEK-293T细胞。Western Blot分析显示,Mpro特征蛋白条带清晰可见,表明经慢病毒转染后的HEK-293T细胞能够持续稳定表达Mpro蛋白(图3A)。在此基础上,对化合物A1至A4开展降解活性评价和作用机制研究。Western Blot实验结果表明,在10 μmol/L浓度下,化合物A4近乎完全降解Mpro(图3B),表现出显著的降解活性,而化合物A1至A3几乎无活性,表明较长的连接子可能有助于提升Mpro PROTAC降解活性。为进一步测定化合物A4的降解活性,我们通过浓度梯度实验结合Image J软件对蛋白条带灰度值进行定量分析,计算得到化合物A4诱导Mpro降解的DC50值为5.2 μmol/L(图3C)。前期在构建质粒时,我们同时插入了编码荧光蛋白(GFP)的基因,以便更直观地观察Mpro的降解情况。用梯度浓度化合物A4处理HEK-293T细胞,孵育24 h后在荧光显微镜下进行观察,结果显示,随着化合物浓度上升,细胞内荧光强度逐渐减弱,表明化合物A4浓度依赖性下调Mpro蛋白水平(图3E),该结果与Western Blot分析一致,进一步验证了化合物A4的降解效果。
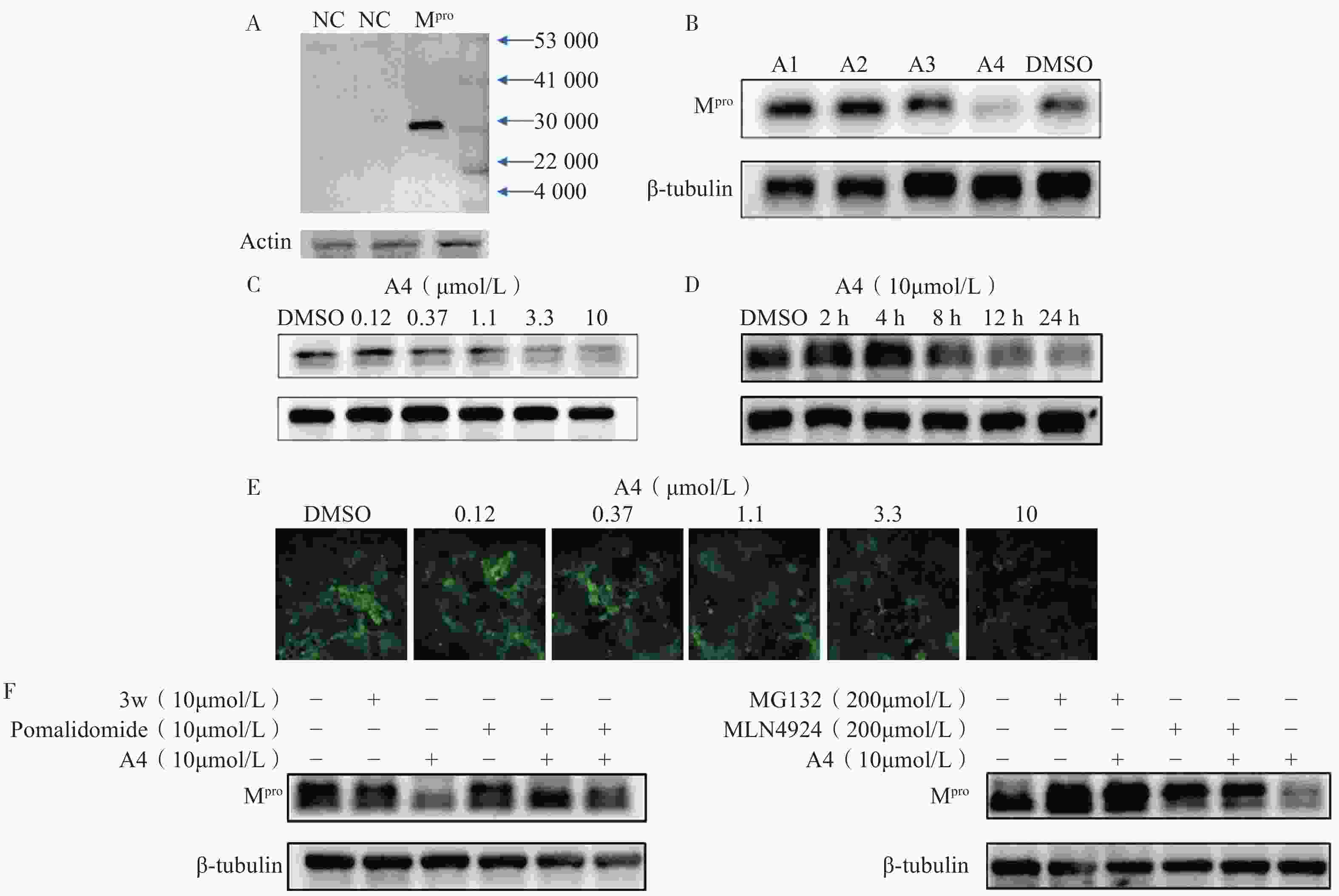
为进一步探究化合物A4的降解动力学特性,我们测定了其在不同孵育时间下导致的Mpro蛋白含量变化(图3D)。结果显示,给药8 h时后Mpro开始降解,24 h达到最大降解效果,表明化合物A4能够以时间依赖性方式诱导Mpro蛋白降解。机制研究表明,在10 μmol/L浓度下化合物A4能够显著下调Mpro蛋白含量。Mpro抑制剂3w或CRBN配体泊马度胺分别与化合物A4共孵育时,均能够阻断化合物A4对Mpro的降解作用(图3F),而两者单独使用时,Mpro蛋白含量未发生改变,表明化合物A4需要两端结合Mpro和CRBN才能发挥相应的降解活性。此外,加入蛋白酶体抑制剂MG132[29]或类泛素化抑制剂MLN4924[30]均可逆转化合物A4对Mpro的降解作用,而单独使用这些抑制剂时,细胞内Mpro的含量不变(图3G)。以上实验结果说明,化合物A4通过与Mpro蛋白和CRBN蛋白的同时结合,诱导Mpro蛋白泛素化修饰并促进其被蛋白酶体降解,从而证明了化合物A4是一种通过泛素-蛋白酶体途径介导Mpro降解的降解剂。
-
基于PROTAC技术,我们设计并合成得到4个靶向SARS-CoV-2 Mpro的PROTAC小分子化合物,所有目标化合物均经过了核磁共振和高分辨质谱确证其化学结构,并通过高效液相色谱确认纯度均达到95%以上。为评估目标化合物对Mpro的降解活性,构建了稳定表达SARS-CoV-2 Mpro的HEK-293T细胞系,并通过Western Blot和荧光显微镜观察验证了Mpro的持续稳定表达。初步筛选结果显示,连接子长度显著影响PROTAC降解效率,其中,连接子最长的化合物A4表现出最优的Mpro降解活性,DC50值为5.2 μmol/L。机制研究进一步证实,化合物A4同时结合靶蛋白Mpro和E3连接酶CRBN,触发泛素-蛋白酶体途径,诱导Mpro蛋白特异性降解。
本研究首次探索了以结构简单的抑制剂3w为Mpro配体的PROTAC分子的设计与生物活性,验证了新型Mpro PROTAC A4在细胞内降解Mpro的有效性,为开发新型抗冠状病毒的PROTAC降解剂提供了先导化合物。尽管化合物A4展现出一定的降解活性,但其DC50值仍有进一步优化的空间,后续的结构优化正在进行中。
Design, synthesis and degradation activity of PROTAC degraders of SARS-CoV-2 main protease
doi: 10.12206/j.issn.2097-2024.202503063
- Received Date: 2025-01-01
- Rev Recd Date: 2025-04-01
- Available Online: 2025-05-12
-
Key words:
- SARS-CoV-2 /
- main protease /
- PROTAC /
- protein degradation activity /
- antiviral drug
Abstract:
| Citation: | WEI Lai, DONG Guoqiang, SHENG Chunquan. Design, synthesis and degradation activity of PROTAC degraders of SARS-CoV-2 main protease[J]. Journal of Pharmaceutical Practice and Service. doi: 10.12206/j.issn.2097-2024.202503063

|
 DownLoad:
DownLoad: