


-
拉莫三嗪是一种苯基三嗪类药物,化学名为6-(2,3-二氯苯基)-1,2,4-三嗪-3,5-二胺,属于第二代抗癫痫药物,临床上常被用于治疗部分性发作癫痫、难治性癫痫、青少年肌阵挛癫痫、癫痫持续状态等[1-4]。拉莫三嗪不经细胞色素P450酶代谢,主要由尿苷二磷酸葡醛酸转移酶(UDP-glucuronosyltransferase,UGT)作用,与葡萄糖醛酸键合,生成无药理活性的代谢物[5]。同其他抗癫痫类药物一样,拉莫三嗪治疗范围(3~14 μg/ml)较窄,易发生中毒反应。
研究拉莫三嗪治疗的446例患者中有20.88%发生不良反应,最常见的有头痛、皮疹、乏力等,严重不良反应有DIC、Lyell综合征、Stevens-Johnson综合征等,研究认为这可能与拉莫三嗪的血药浓度有关[6-7]。因此,随着拉莫三嗪在临床上的广泛应用,治疗药物监测(TDM)成为指导拉莫三嗪合理用药的一种安全有效的手段。本研究旨在通过中心切割二维液相色谱法,建立测定人血浆中拉莫三嗪药物浓度的方法,便于临床开展拉莫三嗪血药浓度监测工作,为临床个体化用药指导提供监测数据支持。
-
Shimadzu LC-20A日本岛津高效液相色谱仪(真空脱气机,分析型色谱泵,SPD双波长检测器);ALK-108二维液相色谱耦合仪(湖南德米特仪器有限公司);色谱柱SNCB(T)-1A(硅胶,4.6 mm ×50 mm, 5 µm)、Symmetry C18(4.6 mm×250 mm, 5 µm)、SBX4-MA(树脂,3.0 mm×10 mm,5 µm)均购自湖南德米特仪器有限公司;QUINTIX125D-1CN精密电子天平(赛多利斯(上海)贸易有限公司);超纯水机(德国Think-lab Corporation);HC-2062高速离心机(安徽中科中佳科学仪器有限公司)。
-
拉莫三嗪标准品(批号:100775-201902,纯度99.9%,中国食品药品检定研究院);乙酸铵(批号:5666364,纯度98.0%,天津迪马科技有限公司);甲醇(色谱纯,上海科丰实业有限公司);乙腈(色谱纯,默克股份两合公司);VCV-1D移动相(湖南德米特仪器有限公司);空白血浆(联勤保障部队第九〇〇医院血库提供);超纯水(实验室自制)。
-
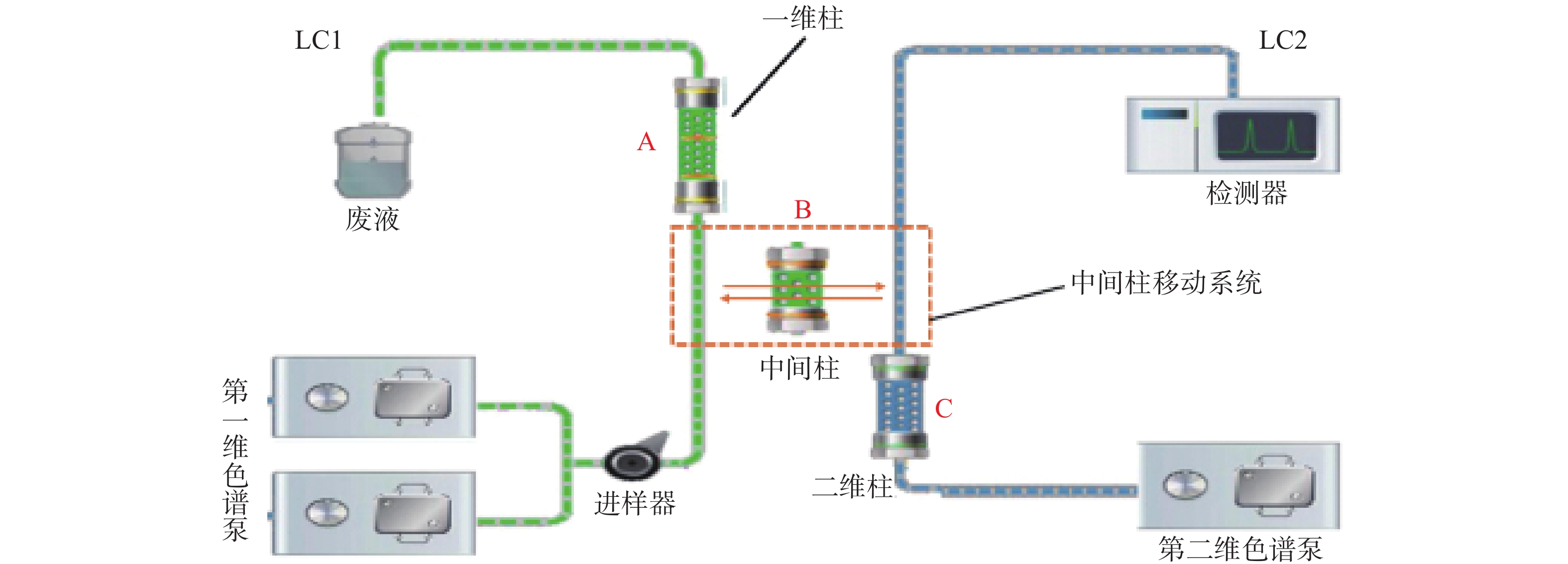
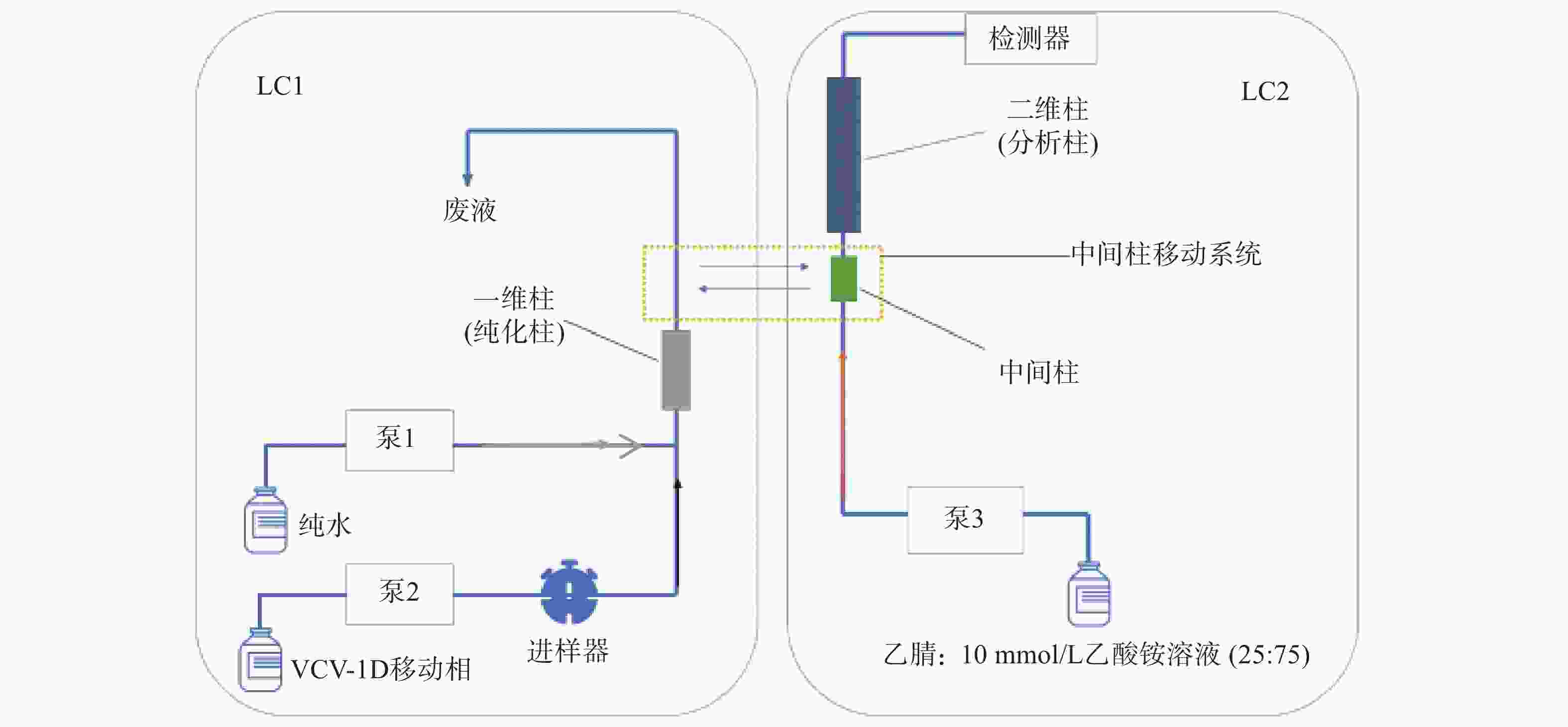
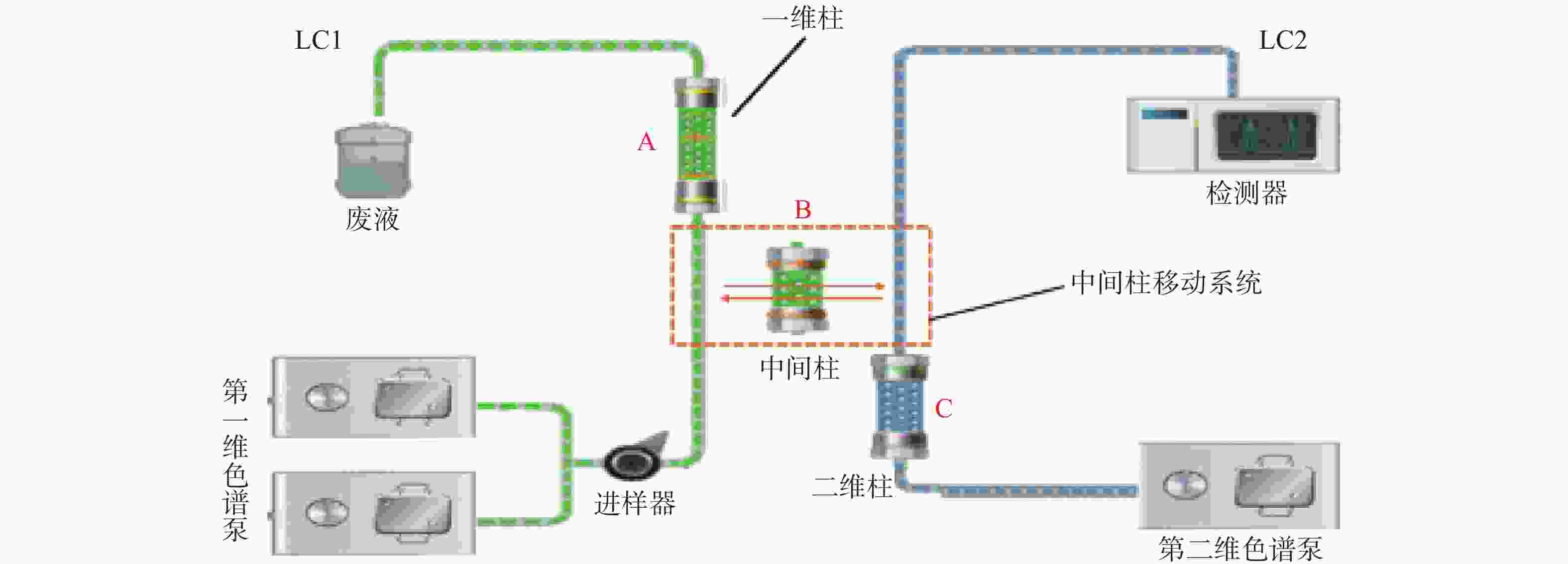
第一维色谱系统中,一维色谱柱:SNCB(T)-1A(硅胶,4.6 mm×50 mm, 5 µm),流动相A:VCV-1D移动相[甲醇-乙腈-磷酸铵水溶液(V/V/V=1∶3∶3,氨水调pH至7.0)],流速:0.4 ml/min;流动相B:水,流速:1.0 ml/min。第二维色谱系统中,二维色谱柱:Symmetry C18(4.6 mm×250 mm, 5 µm),流动相:乙腈:10 mmol/L乙酸铵溶液(25∶75),流速:1.0 ml/min;中间色谱柱:SBX4-MA(树脂,3.0 mm×10 mm,5 µm)。柱温:45 ℃,紫外检测波长:306 nm,进样量:200 μl。时间程序:0~2 min样品进样和第一维色谱分离;2~4 min目标物通过中间色谱柱转移至第二维色谱;4~10 min目标物在第二维色谱中分离分析。
二维液相色谱将分离原理不同的两支色谱柱经一定接口和柱切换技术耦合在一起,通过加入不同的流动相进行选择性分离。样品在第一维色谱进行初步富集分离,经过纯化后将目标组分利用柱切换技术由中间柱转移系统转移到第二维中,最后目标组分在第二维色谱中分离分析。当目标组分在第二维色谱中分离分析时,纯化柱已经逐渐开始清洗,以备样品的再次进入,由此间隔不断地运行。二维液相色谱系统结构见图1;工作原理示意图见图2。
-
精密称取拉莫三嗪标准品7.90 mg,置于100 ml容量瓶中,用甲醇稀释到刻度,摇匀,配制成浓度为79.00 μg/ml的拉莫三嗪标准品储备液,存放在−80 ℃冰箱中备用。
-
精密吸取含药血浆样品200 μl至1.5 ml EP管中,再精密加入甲醇600 μl沉淀蛋白,涡旋混合1 min,4 ℃、12 000 r/min离心10 min,取上清液,进样分析。
-
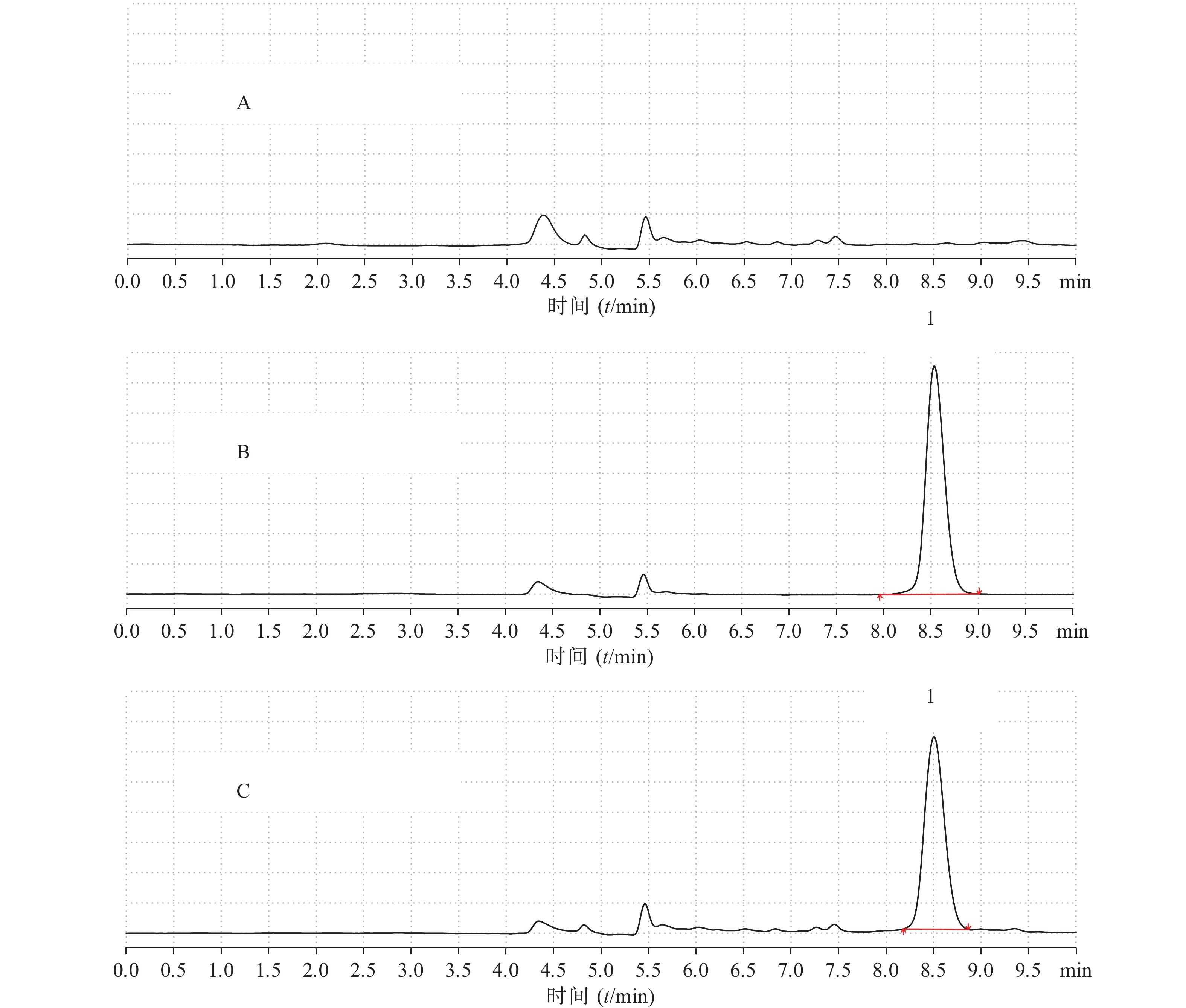
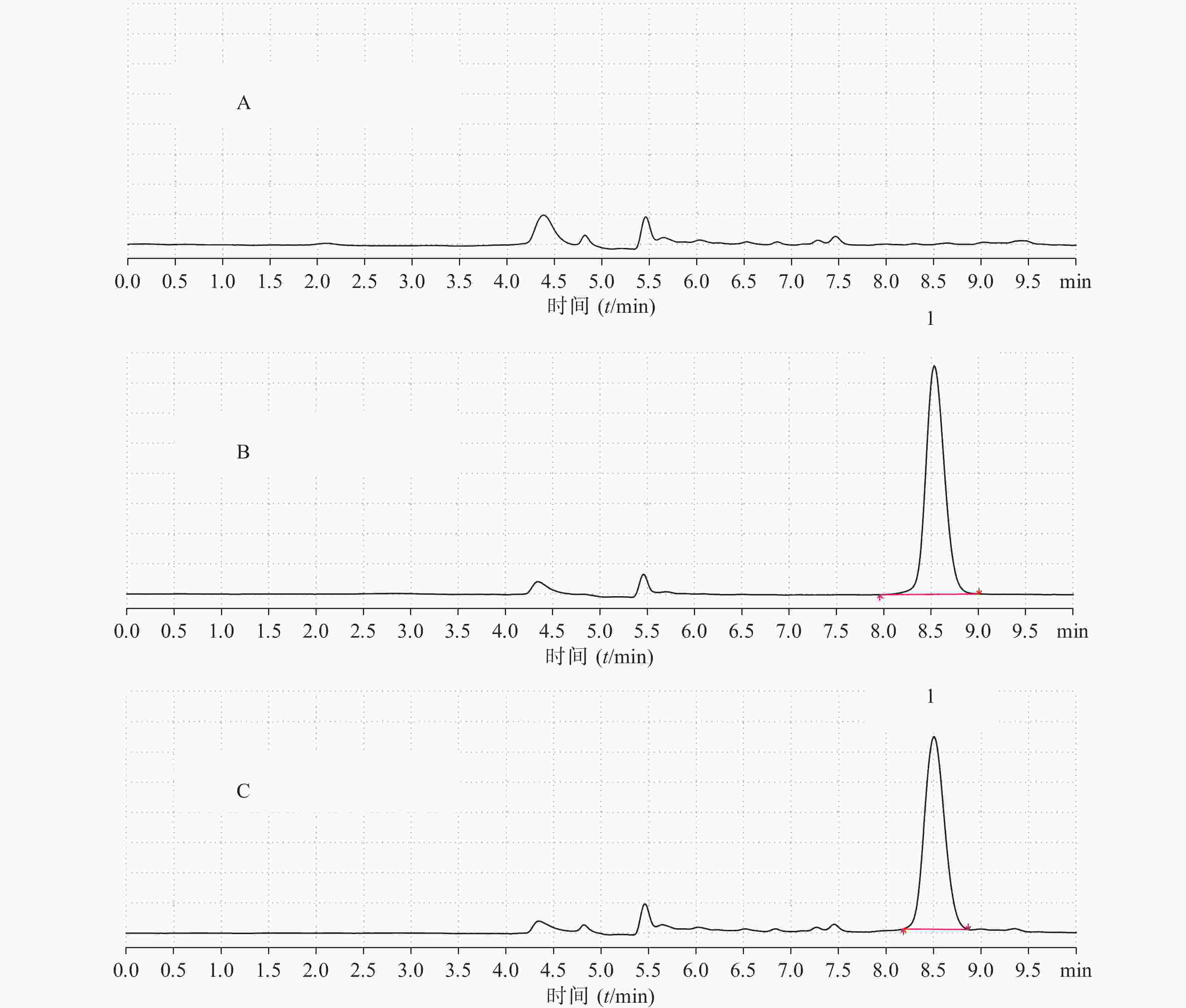
制备空白血浆、空白血浆+拉莫三嗪对照品溶液(浓度为7.90 μg/ml),按照“2.3”项下方法处理,将空白血浆、拉莫三嗪对照品、空白血浆+拉莫三嗪对照品按“2.1”项下色谱条件进样测定,观察有无内源性干扰,如图3所示。结果表明,拉莫三嗪出峰时间为8.506 min左右。血浆中的内源性物质对拉莫三嗪的出峰时间和峰面积无明显干扰(理论踏板数为8 843,对称因子为1.13)。
-
用空白血浆将拉莫三嗪标准品储备液配制成系列浓度梯度溶液(39.50、19.75、9.88、4.94、2.47、1.24 μg/ml),按照“2.3”项下方法处理后,按“2.1”项下色谱条件进样测定。以浓度为横坐标,峰面积为纵坐标进行线性拟合。得到线性回归方程:Y=146 470X-36 830,r=0.999 9,表明拉莫三嗪血药浓度在1.24~39.50 μg/ml时,线性关系良好,定量下限浓度为1.24 μg/ml(S/N=10),检测限为0.02 μg/ml(S/N=3)。
-
用空白血浆将拉莫三嗪标准品储备液配制成浓度为高、中、低(31.60、7.90 、1.98 μg/ml)以及定量下限(1.24 μg/ml)4个浓度的含药血浆样品,每个浓度设置5组平行,按照“2.3”项下方法处理后,按“2.1”项下色谱条件进样测定。1 d内不同时间测定5次及3 d内每天进样1次,考察其日内和日间精密度/准确度。结果如表1~2所示,高、中、低3个浓度的日内RSD均小于5%,日间RSD均小于10%;日内准确度范围为102.17%~111.17%,日间准确度范围为99.80%~107.31%;定量下限浓度的日内、日间RSD均小于5%,日内准确度为100.65%,日间准确度为100.50%。
理论浓度
(μg/ml)测定浓度平均值
(μg/ml)标准差 RSD/准确度
(%)31.60 32.28 0.65 2.02/102.17 7.90 8.27 0.24 2.93/104.66 1.98 2.20 0.06 2.74/111.17 1.24 1.25 0.01 0.58/100.65 理论浓度
(μg/ml)测定浓度平均值
(μg/ml)标准差 RSD/准确度
(%)31.60 31.57 1.34 4.23/99.92 7.90 7.88 0.67 8.54/99.80 1.98 2.12 0.19 9.15/107.31 1.24 1.25 0.01 0.52/100.50 -
用空白血浆将拉莫三嗪标准品储备液稀释成浓度为高、中、低(31.60、7.90、1.98 μg/ml)3个浓度的含药血浆样品,每个浓度设置5组平行,按照“2.3”项下方法处理后,按“2.1”项下色谱条件进样测定,计算回收率。结果如表3所示,回收率RSD均小于5%,其变化范围在89.95%~96.15%之间。
理论浓度
(μg/ml)测定浓度平均值
(μg/ml)标准差 RSD
(%)回收率
(%)31.60 30.03 0.58 1.92 95.04 7.90 7.11 0.30 4.19 89.95 1.98 1.90 0.02 0.90 96.15 -
用空白血浆将拉莫三嗪标准品储备液稀释成浓度为高、中、低(31.60、7.90、1.98 μg/ml)3个浓度的含药血浆样品,每个浓度设置5组平行,按照“2.3”项下方法处理后,按“2.1”项下色谱条件进样测定,分别考察血浆样品室温放置24 h、反复冻融3次以及−40 ℃保存2周的稳定性。结果如表4所示,测定浓度/标准浓度比值范围在87.01%~115.88%。符合《中华人民共和国药典》2020版四部通则中,9012生物样品定量分析方法验证指导原则的要求。
检测条件 药物浓度
(μg/ml)测定浓度平均值
(μg/ml)测定浓度/标示浓度
(%)室温放置24 h 31.60 33.50 106.02 7.90 8.39 106.25 1.98 2.29 115.88 反复冻融3次 31.60 29.29 92.67 7.90 6.87 87.01 1.98 2.04 103.00 −40 ℃保存2周 31.60 31.05 98.25 7.90 7.80 98.70 1.98 2.11 106.73 -
查阅国内外文献可知,目前拉莫三嗪血药浓度测定方法[8-15]有HPLC法、液-质联用法、免疫荧光法。HPLC法测定血药浓度时,须经过复杂的前处理;质谱法的仪器成本和检测成本都较高,且对操作人员要求高,不利于临床推广使用;免疫分析操作简单、速度快,但其检测试剂盒价格昂贵。本研究采用的中心切割二维液相色谱系统是将分离机理不同、互相独立的两根或三根色谱柱以串联方式组合,目标化合物被转移至分析柱后,可对一维柱及中间柱进行在线反向冲洗,可有效减少色谱柱的污染。且与传统HPLC相比,二维液相具有进样体积大(最高可达1 000 µl)、在线净化除杂、富集等功能,可有效避免传统液相色谱测定中因仪器灵敏度和色谱柱分离度的限制,提高临床样本检测的准确性;若质控样品在允许的靶值范围内,则无需每天测定随行标准曲线,可有效缩短临床样本的检测时间,提高仪器的使用率。
高效液相色谱法和液-质联用法大多采用内标法定量,而内标物的选择需考虑其与待测物的物理化学性质是否相似,有时候难以找到合适的内标物;此外,内标物的纯度对定量结果也有很大影响。而二维液相色谱法则采用了外标法定量,在本实验中日内、日间精密度RSD均小于10%,日内准确度的范围为102.17%~111.17%,日间准确度的范围为99.80%~107.31%,以上结果均证明采用外标法是可靠的,不仅实现准确地定量分析,还避免了选取内标的繁琐操作,使得临床上大量样本的检测更具可操作性。除此之外,流动相B还可根据时间程序设定进行自动化调控,早期阶段可增加第一维色谱柱的极性,使待测组分聚集于色谱中,减少损失。一旦样品进入分析阶段,后台即可启动自动清洗系统,自动清洗二维色谱的连接管路、微流控部件与色谱柱,从而确保连续分析期间的系统可靠性,且延长了色谱柱的使用寿命。
-
本实验选用200 µl的进样量,相比传统HPLC法仅几微升的进样量来说,二维液相色谱法可在线处理大体积进样,实现样品的在线浓缩与富集,有效消除基质干扰,且二维液相色谱法具有的全息反射长光程检测技术可将检测器提高1.2~1.8倍,使得灵敏度大幅度提高;二维液相色谱法还利用双级流体调制控制技术,巧妙的通过辅助流动相-水在线调制一维流动相-VCV-1D移动相,改变流动相特性、提高分离能力;本次实验过程中,通过组合调配理化性质不同的色谱柱,利用其分离能力的不同,增加色谱柱对目标物质的选择性,精准控制目标物质的迁移。对比其他色谱法,二维液相色谱法的分离能力大幅度提高[16]。
-
目前常用的生物样品前处理方法有蛋白沉淀法、液-液萃取法和固相萃取法。液-液萃取法以及固相萃取法过程复杂且成本较高[17-18],而蛋白沉淀法能够最大限度地保留样品中的物质,操作简单且成本较低[19],其中,有机溶剂沉淀法是蛋白沉淀法最常用的方法。血浆或血清样品常采用甲醇或乙腈沉淀蛋白,样品与沉淀剂比例为乙腈最低1∶2,甲醇最低1∶3(通常1∶5以上效果最佳)。本研究实验样品为血浆,且配置标曲所用的溶剂为甲醇,为了避免不同有机试剂对检测的干扰,因此选择甲醇为沉淀试剂,样品与甲醇的比例为1∶3。
-
预实验时采用的一维色谱柱是SNCB-1A色谱柱,固定相为硅胶,当含药血浆样品通过色谱柱时,其中的各组分会因其与固定相的亲和力差异而发生不同的吸附或扩散现象,与湖南德米特公司提供的配套试剂VCV-1D移动相互相作用,能实现在线萃取聚焦功能,极大地简化了前处理过程,提高仪器的灵敏度,其结果分离度良好,有较好的峰型,且无拖尾现象,采用“中心切割技术”只将一维中洗脱出来的目标组分切割后再转入第二维分析。而二维色谱柱采用的是非极性的C18柱,进行洗脱时选择性差异取决于不同pH下的溶解性。拉莫三嗪在水中不易溶解,但在氯仿、二氯甲烷等有机溶剂中易溶解。目前常用的流动相包括甲醇-水、乙腈-水、乙腈-磷酸盐缓冲液等。参考以上流动相进行预实验,拉莫三嗪并不能完全分离分析。考虑到拉莫三嗪是一种弱碱性的药物,在酸性条件下易于质子化,而在碱性条件下易于去质子化。因此,为了保证其在分离过程中的稳定性和分离效果,需选择适当的缓冲液进行调节;又因拉莫三嗪分子结构中含有芳香环,具有一定的极性,则需选择一定极性的流动相来实现其与固定性的相互作用,从而实现有效分离。经多次实验后,确定二维流动相(乙腈-乙酸铵)比例为25:75,在此条件下拉莫三嗪具有良好的峰型且无拖尾现象。
综上考虑,本研究选用色谱柱SNCB-1A、Symmetry C18作为二维液相色谱法的第一维色谱柱、第二维色谱柱,VCV-1D移动相为第一维流动相,乙腈-乙酸铵溶液为第二维流动相。
-
相比于普通液相的进样量,二维液相色谱的要大十几倍,主要是因为一维色谱柱的柱长比较短,仅50 mm,因此,柱压不会因为较大的进样量而明显增加。除此之外,还有流动相B的洗脱作用,可使待测样品在色谱柱上富集,故当进样量增加时,峰型也不受影响。本实验中进样量采用200 μl,是基于定量限需满足临床血药浓度的测定要求来确定的。若还需提高检测灵敏度,进样量最高可以达到1 ml。
-
本研究建立的中心切割二维液相色谱测定方法操作简单、稳定性好、灵敏度高、重现性好,便于临床开展拉莫三嗪血药浓度监测工作,可为临床个体化用药指导提供可靠的监测数据支持。
Determination of Lamotrigine in Human Plasma by Central cutting two-dimensional liquid Chromatography
doi: 10.12206/j.issn.2097-2024.202306020
- Received Date: 2023-06-12
- Rev Recd Date: 2024-03-17
-
Key words:
- lamotrigine /
- plasma concentration /
- two-dimensional liquid chromatography /
- central cutting
Abstract: Objective To establish a central cutting two-dimensional liquid chromatography for the determination of lamotrigine in human plasma. Methods External standard method was used. The first dimensional chromatographic column: SNCB(T)-1A(silica gel, 4.6 mm×50 mm, 5 μm), mobile phase A:VCV-1D mobile phase, flow rate: 0.4 ml/min; mobile phase B: water, flow rate: 1.0 ml/min; second dimensional chromatographic column: Symmetry C18 (4.6 mm×250 mm, 5 μm), mobile phase: acetonitrile-10 mmol/L ammonium acetate solution(V/V=25∶75), flow rate: 1.0 ml/min; Intermediate column: SBX 4-MA(resin, 3.0 mm×10 mm, 5µm). The UV detection wavelength: 306 nm, the column temperature: 45 ℃, and the injection volume: 200 μl. Results The linear range of lamotrigine was 1.24-39.50 μg/ml, the lower limit of quantification was 1.24 μg/ml, the detection limit was 0.02 μg/ml, the intra-day precision RSD was less than 5%, the day-to-day precision RSD was less than 10%, the variation of intra-day accuracy ranged from 102.17% to 111.17%, and the daytime accuracy variation ranged from 99.80% to 107.31% the recovery RSD was less than 5%, and the variation range was 89.95% -96.16%. After 24 hours storage at room temperature, repeated freezing and thawing for 3 times and storage at-40 ℃ for 2 weeks, the ratio of the measured value / labeled value ranged from 87.01% to 115.88%. Conclusion In this study, a method with simple operation, good stability, high sensitivity and good reproducibility was established, which could be suitable for clinical monitoring of blood concentration of lamotrigine and provides reliable monitoring data support for clinical individualized medication guidance.
| Citation: | LIN Junrong, ZHANG Minxin, ZHAO Xuemei, HUANG Aiwen. Determination of Lamotrigine in Human Plasma by Central cutting two-dimensional liquid Chromatography[J]. Journal of Pharmaceutical Practice and Service. doi: 10.12206/j.issn.2097-2024.202306020

|
 DownLoad:
DownLoad: